Introduction
Many biomolecules contain a phosphate group in their structure, whose presence plays a crucial role in the functioning of nucleotides, nucleic acids, enzymes, transporters, sugars, lipids, etc. Easy access to phosphorylated derivatives is of paramount importance in many studies. Thus, phosphorylation has attracted the attention of many chemists and biochemists of late, resulting in several new approaches. Chemical methods in phosphorylation can be divided into two general categories depending on whether P(III) or P(V) phosphorylating agents are used. P(III) phosphorylating agents offer a significantly higher reaction rate (sometimes the reactivity is too high), but they require an additional step of oxidation. On the contrary, P(V) phosphorylating agents introduce the phosphorus atom in its final oxidation state, but the reactions are often slow. An additional factor that has to be taken into account during phosphorylation is the use of phosphorus-protecting groups, which are often necessary due to the trifunctional character of phosphorus acid (Kraszewski and Stawinski, 2002).
Phosphorylation strategies using P(III) agents are classified into two major groups: phosphitylation and phosphonylation, both followed by oxidation. The former uses tervalent phosphorus compounds, e.g., PCl3 (Beni et al., 2015; Brusentsev and Eklund, 2016), phosphoro[mono/di]chloridites (Letsinger et al., 1975; Mara et al., 2011; Stowell et al., 1995), phosphoro[mono/di]amidites (Beaucage and Caruthers, 1981), and phosphorochloridoamidites (Beni et al., 2015). These reactants are usually highly reactive, particularly in the presence of bases (P–Cl compounds) or acids (P–N compounds), and are moisture and air sensitive. In phosphonylation, H3PO3 and its mono- and diesters (H-phosphonates) are used as the sources of phosphorus. They are stable compounds – they neither hydrolyze nor undergo oxidation by air when stored on the shelf. For reactions with nucleophiles, they need condensing agents (usually pivaloyl chloride), basic conditions, and a nucleophilic catalyst, typically pyridine (Kraszewski and Stawinski, 2007). However, a reverse order of the reaction steps – an initial P(III) → P(V) oxidation followed by a reaction with a nucleophile – is also possible. To this end, phosphite triesters (Arbuzov-type reaction) or H-phosphonate diesters (Atherton–Todd reaction) are treated with a halogenating agent (e.g., Br2, I2, CCl4) to form halogenophosphate diesters that function as phosphorylating reagents (Della-Felice et al., 2022).
In the second category of phosphorylation strategies, phosphorus acid derivatives [P(V) compounds] are used. Similarly as for P(III) compounds, reactive P-[LG] derivatives can be used, e.g., P(O)Cl3 (Jakubovska et al., 2018) or pyrophosphates (Dixit et al., 1981). Alternatively, inert phosphate esters can be activated using various condensing agents and applied in situ for phosphorylation (Della-Felice et al., 2022).
Della-Felice et al. (2022) have recently reviewed several phosphorylation methodologies. Despite the large number of known approaches, new phosphorylation strategies are being developed, showing a steady demand for phosphorylated compounds. In this paper, the synthetic potential of an unusual class of compounds recently obtained in our laboratory, namely oxyonium phosphobetaines (Materna et al., 2016), was discussed.
Materials and methods
Chemicals
The reaction conditions and procedures for preparation of reactants and solvents are based on a previous study (Materna et al., 2016).
General procedure for phosphorylation of protected nucleosides
A suitably protected nucleoside (0.5 mmol) was rendered anhydrous by repeated evaporation of the added anhydrous pyridine and then toluene, and left under vacuum overnight. An N-oxide (6 equiv.) was also kept under vacuum overnight. Then, the nucleoside was dissolved in 7 : 3 dimethylformamide (DMF)/pyridine (Py) (v/v, 5 ml), and activated powdered molecular sieves 4 Å were added, followed by diphenyl H-phosphonate (DPP, 2.5 equiv.; reaction mixture A). The mixture was stirred using a magnetic stirrer for 15 min (for phosphonylation of 5′−OH compounds) or 20 min (for phosphonylation of 3′−OH compounds). Meanwhile, the N-oxide was dissolved in the same solvent system (5 ml) at 40°C, with the addition of activated powdered molecular sieves 4 Å. This solution was added to the reaction mixture A and left for 10 min. Then, the flask was kept in a freezer (−18°C) overnight. The precipitated phenyl oxyonium phosphobetaine 3 (G = Ph) was removed by decantation. Occasionally, if the residue was too labile for decantation, it was isolated by filtration. Triethylamine (4 equiv.) and trimethylsilyl chloride (TMSCl, 11 equiv.) were added sequentially while stirring. After 5 min, an excess of methanol (10 ml) was added, and the mixture was concentrated in a rotary evaporator. The oily residue was partitioned between water and DCM. The aqueous phase was extracted with DCM, and the combined organic phases were evaporated with a few drops of triethylamine to prevent detritylation. The residue was dissolved in a small volume of methanol and precipitated from diethyl ether to yield an amorphous product. If the product was contaminated by orange impurities, precipitation was repeated. Finally, the product was purified by silica gel chromatography using a 0 → 10% gradient of water in a 95:5 (v/v) acetonitrile-triethylamine solvent system. After evaporation, the product was obtained as a white amorphous powder.
Results and discussion
During our studies on new phosphorylation strategies, we observed that H-phosphonate esters with a good leaving group reacted rapidly with amine N-oxides with the formation of previously unknown compounds – oxyonium phosphobetaines 3 having a zwitterionic structure with a unique -O–P–O–N+ bond system. A putative mechanism involving a sequence of two successive reactions with two molecules of an N-oxide is represented in Scheme 1.
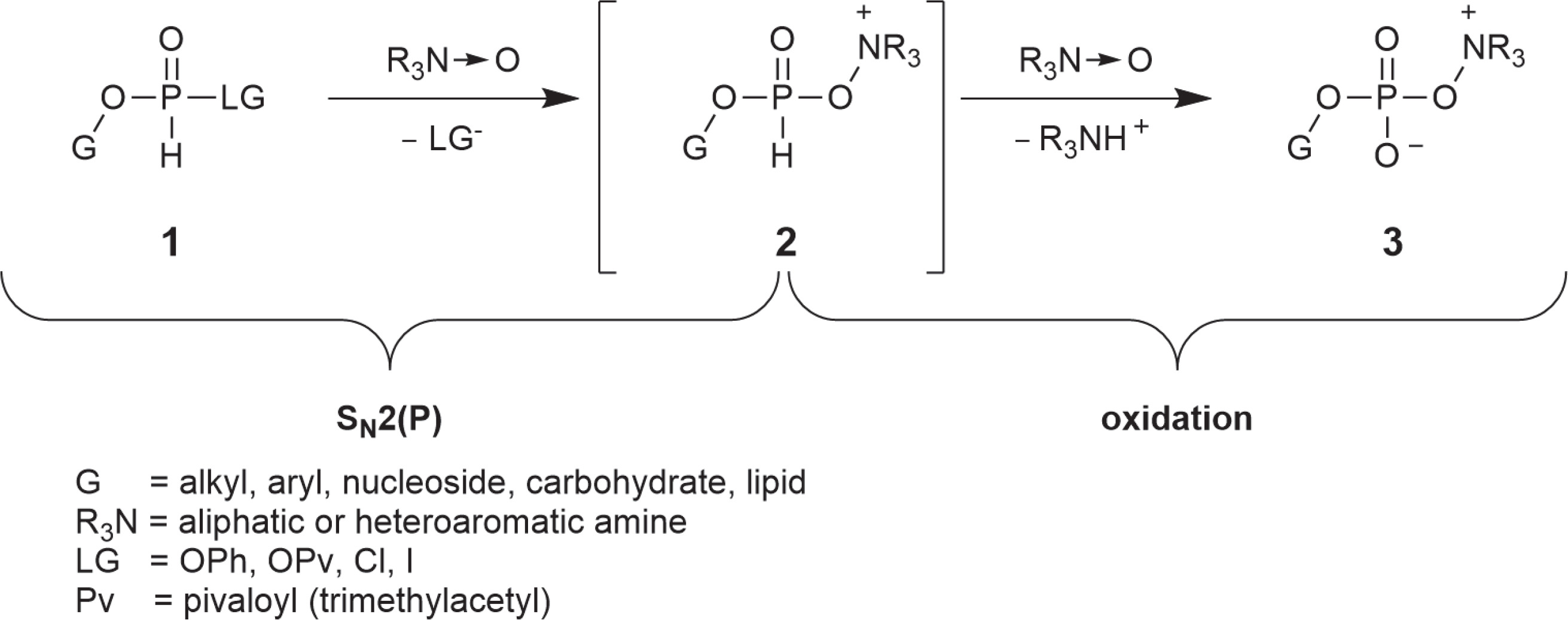
Oxyonium phosphobetaines 3 were formed efficiently and appeared to be surprisingly stable, and many of them could be isolated and stored. On the other hand, they reacted easily with various nucleophiles, which made it possible to develop a new approach to obtaining various biologically important phosphate derivatives (Scheme 2).
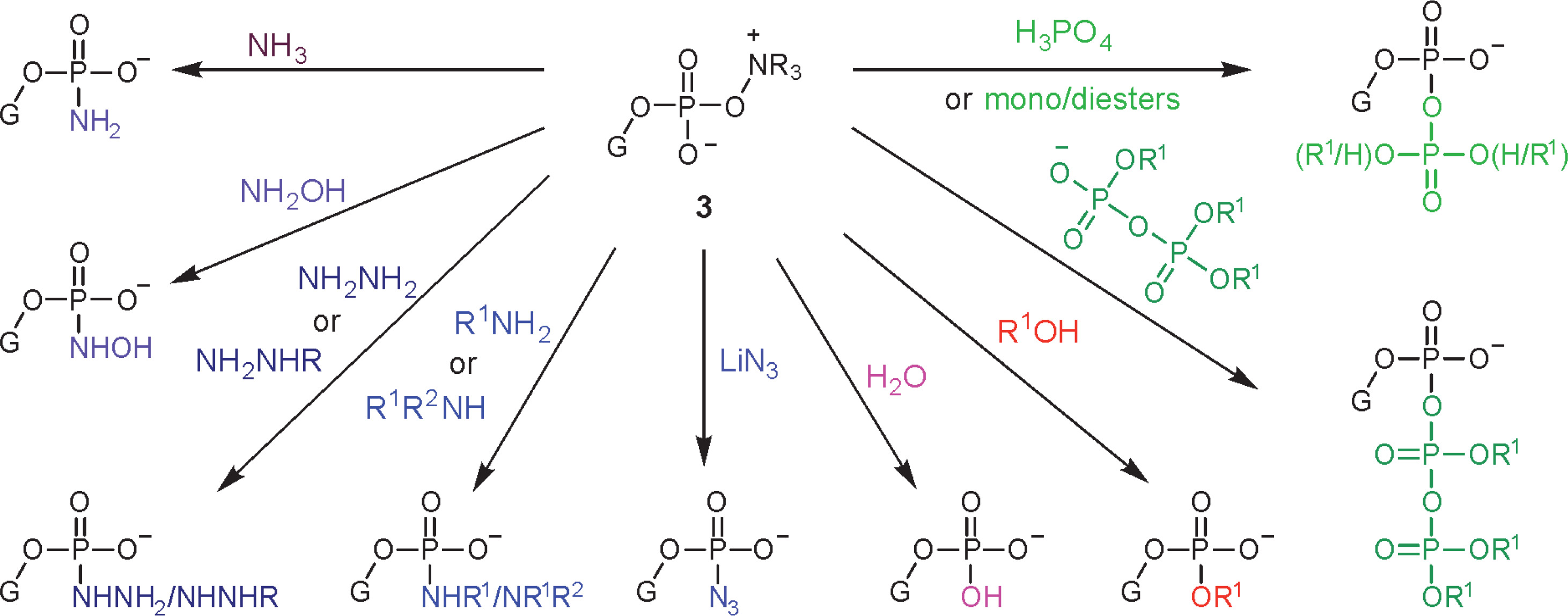
The scope of N-oxides that can be used in phosphorylation is broad and includes N-oxides of aliphatic (trimethylamine, N-methylmorpholine) and heteroaromatic amines (pyridine and its substituted derivatives). In the studies reported in this paper, appropriately protected nucleosides 4 were converted into nucleoside phenyl H-phosphonate diesters 5 and treated with N-methylmorpholine N-oxide (NMMO, an N-oxide of choice, vide infra), resulting in oxyonium phosphobetaines 6. These intermediates could be isolated for their characterization or directly converted into final products (Scheme 3). A procedure for the preparation of monophosphates 7 (Z = OH) was presented in this paper, and the methods for obtaining other derivatives represented in Scheme 2 will be published in due course.
Scheme 3
A three-step, one-pot procedure of phosphorylation of nucleosides (4) toward nucleotides or their derivatives (7) via oxyonium phosphobetaines (6); the same sequence of reactions can be used for the phosphorylation of the 5′−OH group of nucleosides; the scope of Z is shown in Scheme 2
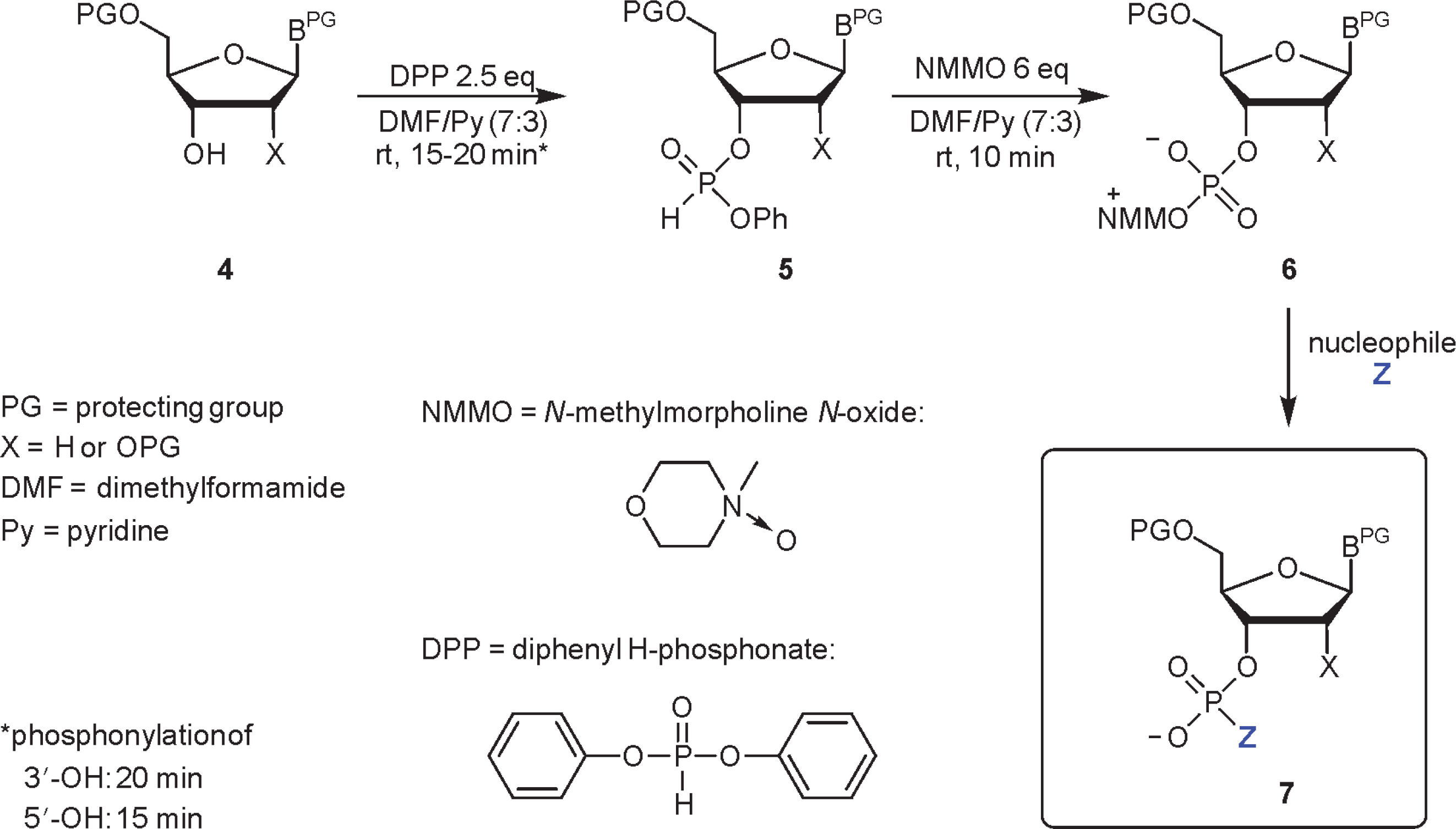
H-phosphonate diesters of type 1 were obtained in situ using a slightly modified transesterification approach reported by Jankowska et al., i.e., by treating the nucleoside with DPP (Jankowska et al., 1994). In the original method, three molar equivalents of DPP with respect to the nucleoside were used to prevent double transesterification and formation of dinucleoside H-phosphonate. In this paper, excess of DPP was unfavorable since it also reacts with N-oxides to produce phenyl betaines of type 3 (Scheme 1, G = Ph), and this reaction could interfere with the final step of the procedure and complicate the isolation of products. Unfortunately, it was not possible to decrease the amount of DPP to less than 2.5 equiv. without deterioration of the yield of nucleoside phenyl H-phosphonate 5. The solvent used was a 7 : 3 (v/v) mixture of DMF-Py, in which the formation of dinucleoside H-phosphonate was slower and N-oxides were dissolved efficiently in subsequent steps. Since reducing the basicity of the solvent could decrease the rate of formation of the desired intermediates of type 5, the progress of the reactions was monitored using 31P NMR spectroscopy. The complete phosphonylation of the 5′−OH function of nucleosides required 15 min, whereas for those with free 3′−OH, 20 min was required. Since aryl H-phosphonate diesters of type 5 are moisture sensitive and prone to disproportionation under basic conditions (Kers et al., 1996), they need to be prepared immediately prior to the reaction with an N-oxide.
In the next step, i.e., the generation of nucleoside oxyonium phosphobetaines of type 6, four N-oxides were tested: two aliphatic [trimethylamine N-oxide and NMMO] and two aromatic [4-methoxypyridine N-oxide and 4-(N,N-dimethylamino)pyridine N-oxide]. Their reactions with H-phosphonates of type 5 (Scheme 2) derived from two nucleosides (one with a free 3′−OH and one with a free 5′−OH group) were analyzed using 31P NMR spectroscopy (Table 1). In both cases, the best results were obtained for the reaction with NMMO, and thus, this N-oxide was used in further experiments. The advantage of NMMO over other N-oxides tested was confirmed in several other reactions (data not shown).
Table 1
Yields (31P NMR) of the formation of oxyonium phosphobetaines of type 6 [%] from H-phosphonate diesters of type 5 and N-oxides (conditions as in Scheme 3); the yields were calculated by the integration of the signal of product 6 and the nucleosidic products in 31P NMR spectra; TMAO – trimethylamine N-oxide, NMMO – N-methylmorpholine N-oxide, MEONO – 4-methoxypyridine N-oxide, DMAPO – 4-(N,N-dimethylamino)pyridine N-oxide, 5′-DMTrT – 5′-O-(4,4′-dimethoxytrityl)thymidine, AZT – 3′-deoxy-3′-azidothymidine
| N-oxide | 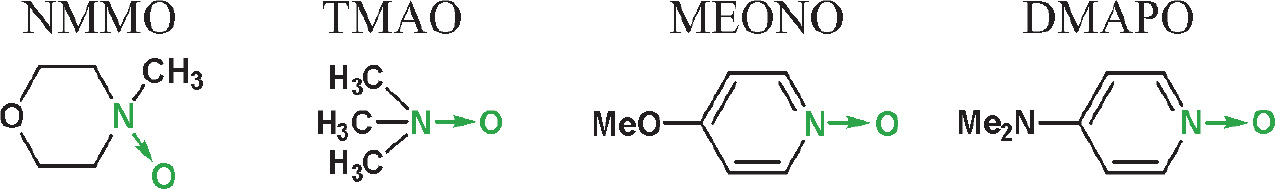 | |||
|---|---|---|---|---|
| Nucleoside 4 | ||||
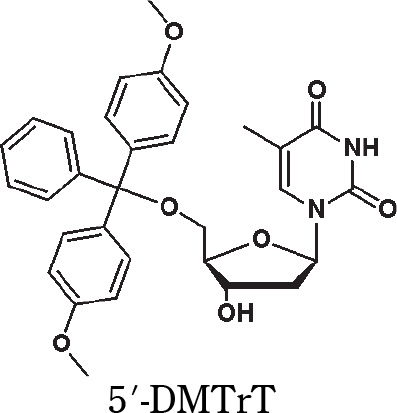 | 100 | 100 | 23 | 93 |
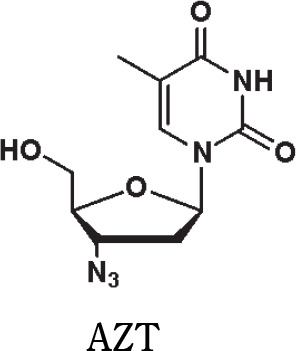 | 100 | 86 | 48 | 97 |
In the majority of the cases, the nucleoside oxyonium phosphobetaines obtained were stable enough to be isolated. However, chromatographic removal of phenyl oxyonium phosphobetaine, which was formed as a side product from DPP and N-oxide, was extremely difficult.
Most of this side product precipitated from the reaction mixture spontaneously, whereas the remaining was removed using a simple additional procedure. To this end, the reaction mixture was filtered, and the precipitate was washed with toluene. Diethyl ether was added dropwise to the combined solutions, and the mixture was kept for 1 h at 0°C. This resulted in the precipitation of practically all phenyl oxyonium phosphobetaine, whereas nucleoside oxyonium phosphobetaine remained in the solution. After work-up with an aqueous solution of sodium bicarbonate, the organic phase was dried using anhydrous Na2SO4 and evaporated to dryness. The residue was dissolved in dichloromethane (DCM), and the product was isolated using precipitation with diethyl ether. The yields of exemplary nucleoside N-methylmorpholine oxyonium phosphobetaines are presented in Table 2.
Table 2
Yields of nucleoside N-methylmorpholine oxyonium phosphobetaines of type 6; Thy – thymine, CytBz – N 4-benzoylcytosine, AdeBz – N 6-benzoyladenine, Gibu – N 2-isobutyrylguanine, Ura – uracil, TBDMS – tert-butyldimethylsilyl
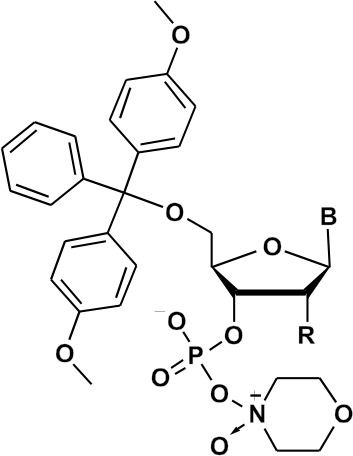
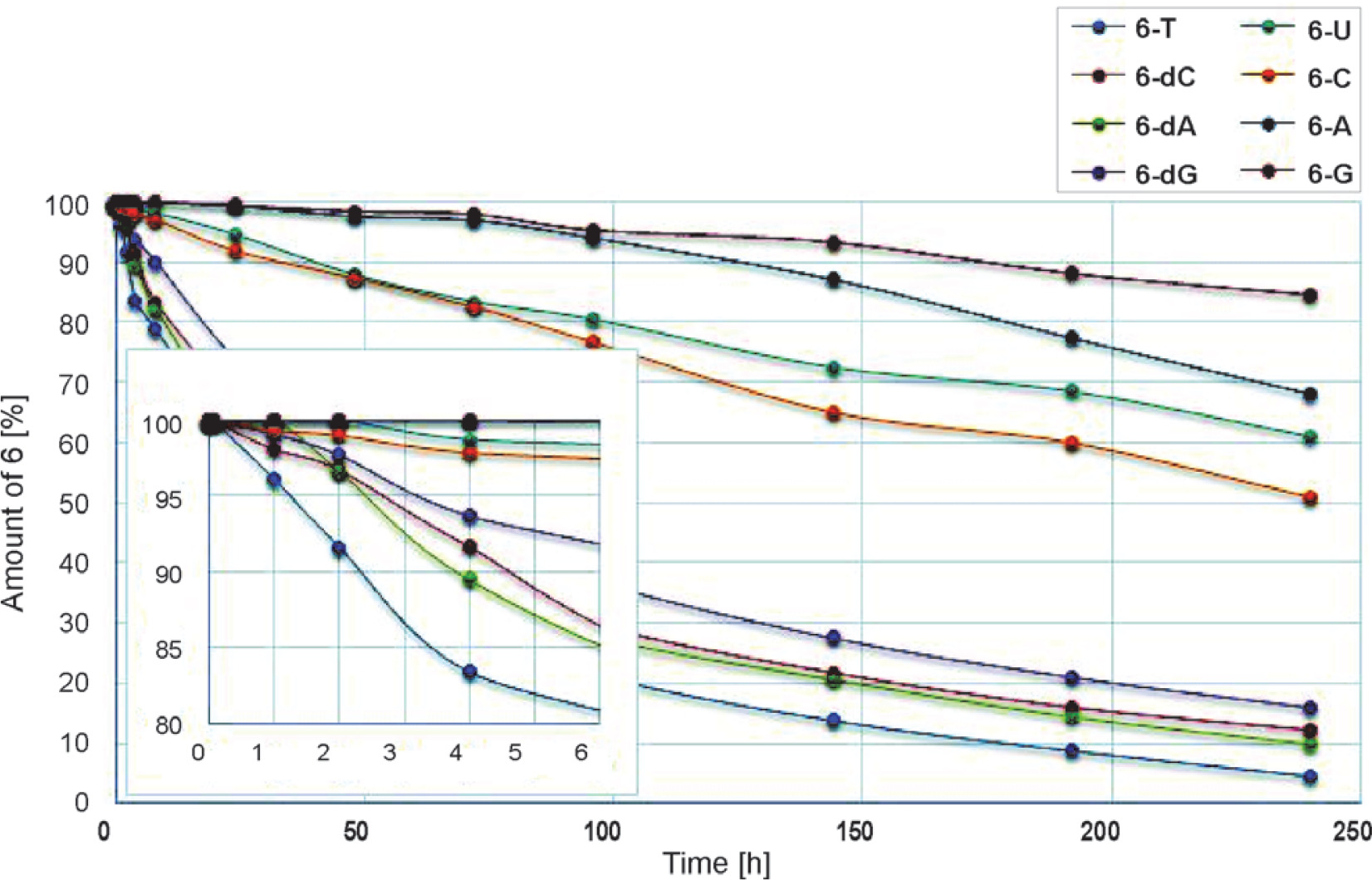
Nucleoside oxyonium phosphobetaines are resistant to hydrolysis. Interestingly, this reaction was much slower in the ribo series than in the deoxyribo one (Fig. 1). This could be due to the steric protective effect of a bulky tert-butyldimethylsilyl group in the 2′-O position.
Fig. 1
Hydrolysis of phosphobetaines 6 in the DMSO/H2O (85 : 15, v/v) mixture (31P NMR). The inset shows the first 6 h of the reactions. Structures are provided in Table 2
Instead of isolating phosphobetaine intermediates of type 6, they can be in situ converted to the desired phosphates 7. Since the hydrolysis rate of compound 6 is low (Fig. 1), simple phosphates (7, Z = OH, Scheme 3) can be synthesized more conveniently using a rapid and efficient silylation/desilylation procedure as shown in Scheme 4 (a putative mechanism). Interestingly, the reactions of this protocol do not require the addition of water. Thus, the reaction mixture containing nucleoside phosphobetaines 6 was kept overnight in a freezer (−18°C), and the precipitated phenyl oxyonium phosphobetaine was removed by decantation (or filtration if decantation was not efficient).
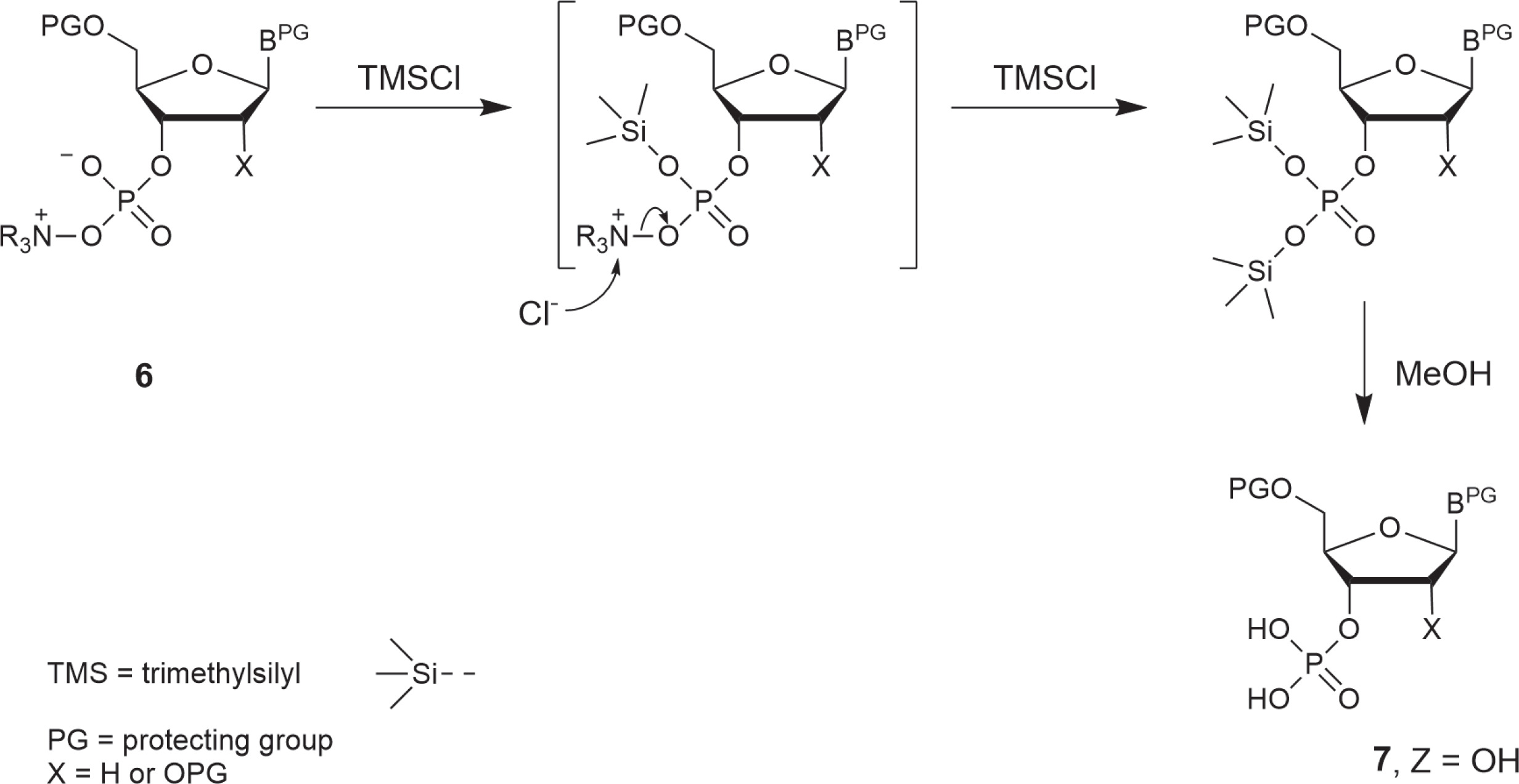
Then, triethylamine (3 equiv.) and TMSCl (11 equiv.) were added, followed by methanol (10 equiv.) after 5 min. The conversion of betaines 6 into phosphates 7 was quantitative, as observed from the 31P NMR spectra. Then, the reaction mixture was fractionated in an H2O–DCM system. To prevent detritylation, a few drops of triethylamine were added to the solution at each step. Nucleoside phosphates were precipitated with diethyl ether and purified using silica gel chromatography. The isolated yields are presented in Table 3. The yields for A, C, and T/U nucleosides were high to very high (70–96%), whereas those for guanosine derivatives were slightly lower (40–47%). Phosphates of type 7 were obtained as triethylammonium salts, which had a purity higher than 95%. Their analytical data (31P, 13C, and 1H NMR spectra) were in agreement with those from the literature (Greenberg et al., 1998; Cieslak et al., 2002).
Table 3
Isolated yields of nucleoside phosphates of type 7 obtained from ribo- and deoxyribonucleosides via oxyonium phosphobetaines 6; abbreviations are presented in Table 2
Yields of nucleoside phosphates 7 | |||
|---|---|---|---|
| Cpd. | B | R | Isolated yield [%] |
| 7-T | Thy | H | 94 |
| 7-dC | CytBz | H | 85 |
| 7-dA | AdeBz | H | 96 |
| 7-dG | GuaiBu | H | 47 |
| 7-U | Ura | O–TBDMS | 86 |
| 7-C | CytBz | O–TBDMS | 70 |
| 7-A | AdeBz | O–TBDMS | 71 |
| 7-G | GuaiBu | O–TBDMS | 40 |
Conclusions
A new procedure for the synthesis of nucleoside phosphates was developed. This preliminary study demonstrates the suitability of the developed protocol for the efficient and rapid synthesis of protected nucleoside phosphates (nucleotides). It is worth noting that the key intermediates, the nucleoside oxyonium phosphobetaines, are universal precursors for numerous phosphate compounds. The developed phosphobetaine derivatives can be isolated and/or used to synthesize various phosphate analogs, e.g., di- and triphosphates, phosphate diesters, phosphoramidates, phosphohydrazidates, phosphorazidates, etc. These procedures will be reported in due course.