






Introduction
Liver fibrosis is a regenerative process resulting from chronic liver damage, which is manifested with the deposition of excess extracellular matrix proteins, posing a global health challenge owing to its increasing prevalence and a lack of effective therapies [1]. Hepatic stellate cells (HSCs), a mesenchymal cell population constituting 5-10% of all cells in the liver, are the predominant cell type responsible for fibrosis [2]. Senescent HSCs are found in liver fibrosis, with reduced collagen production and proliferation [3]. HSCs are activated upon liver damage and can be differentiated into myofibroblasts and proliferated in this process; also, they release the collagen-rich extracellular matrix (ECM) during the process of liver fibrosis [4]. Furthermore, HSC activation is a well-known key driver of liver fibrosis, and understanding its underlying mechanisms has made a remarkable contribution to novel therapeutic strategies for chronic liver disease [5]. Ferroptosis is a non-apoptotic, iron-dependent type of regulated cell death, and the ferroptosis of HSCs may suppress liver fibrosis by removing the main source of collagens [6]. Therefore, targeting HSC activation depending on ferroptosis may aid in the development of new therapies for patients with liver diseases such as fibrosis.
Stem cells have shown promise as novel therapies for multiple incurable chronic diseases, including liver fibrosis. More importantly, mesenchymal stem cells (MSCs), which can secrete a mass of molecules outside cells, such as extracellular vesicles (EVs; including microvesicles and exosomes) and lipids, play critical roles in restoring damaged tissues [7, 8]. Animal experiments have shown the anti-apoptotic, anti-inflammatory and anti-fibrotic capabilities of EVs sourced from diverse MSC types in acute or chronic liver disease [9]. For instance, exosomes (Exos) released by human bone marrow-derived MSCs (BMSCs-Exos) can ameliorate CCl-evoked liver fibrosis by limiting HSC activation [10]. A more recent study demonstrated that MSC-Exos contribute to protection against ferroptosis in mice by modulating SLC7A11 [11]. SLC7A11, a known factor related to ferroptosis, may contribute to the synthesis of glutathione, thus ameliorating the stress-caused injury resulting from lipid reactive oxygen species (ROS) and strengthening the anti-ferroptosis capability of cells [12]. Several studies have indicated a relationship between microRNA (miR)-144-3p and SLC7A11, an important mechanism in regulating cell biological processes such as inflammation and ferroptosis [13-15]. miRNAs, short noncoding RNAs (~22 nucleotides) that control gene expression posttranscriptionally, have the ability to mediate the activation of HSCs [16, 17]. Given that Exos are important carriers of molecules such as miRNAs and proteins, we speculated whether BMSCs-Exos could deliver miR-144-3p to regulate SLC7A11, thus affecting the ferroptosis and activation of HSCs.
Material and methods
Cell culture
Both human BMSCs (HUM-iCell-s011) and LX-2 cells (iCell-h128) were procured from Cellverse Bioscience Technology Co., Ltd. (Shanghai, China). In a 37°C and 5% CO2 incubator, BMSCs were cultured in DMEM/F12 medium that contained 10% fetal bovine serum (Gibco, Grand Island, NY), and LX-2 cells were incubated in DMEM medium (Gibco) harboring 10% FBS replenished with 1% penicillin/streptomycin.
Identification of BMSCs
Bone marrow-derived mesenchymal stem cell culture medium was renewed every 3 days, and cells are harvested after multiple digestions. When the cell confluence reached 80%, they were passaged. Passage 3 cells were harvested and their morphology and growth were observed. Then, the BMSC-positive (CD29, CD90, and CD44) and -negative (CD45 and CD34) markers were detected on a flow cytometer (BD Biosciences, San Jose, CA).
Additionally, the adipogenic, osteogenic, and chondrogenic abilities of the aforementioned cells were identified using alizarin red staining solution (GP1055, Servicebio, Wuhan, China), oil red O staining solution (BP-DL101, SenBeiJia Biological Technology Co., Ltd., Nanjing, China), and Alcian Blue solution (BP-DL241, SenBeiJia). Only passage 3 cells were chosen for subsequent experiments.
Extraction and identification of BMSCs-Exos
The extraction of BMSCs-Exos was carried out according to the description in previous literature [18]. Specifically, BMSCs were seeded in a six-well plate containing MSC growth medium (1 × 105/well). After 24 hours, DMEM medium was supplemented with 10% Exos-free serum (iCell-0650, Cellverse Bioscience, Shanghai, China) for an additional 48 hours. The cultured supernatant was collected, followed by 10-min centrifugation at 4°C and 300 g, 20-min centrifugation at 1000 g, and 30-min centrifugation at 10,000 g. The supernatant was collected and filtered through a 0.22 µm membrane. The precipitates were subjected to ultracentrifugation at 100,000 g for 60 min and resuspension in PBS, followed by another 60-min ultracentrifugation at 100,000 g to eliminate contaminated proteins. Finally, the precipitates were resuspended in PBS and stored at –80°C.
Exos were identified by analyzing their particle size with the Nanosight LM10 system (Nanosight Ltd, Navato, CA), observing their morphology under a transmission electron microscope (H7650, HITACHI, Tokyo, Japan), and detecting the expression of marker proteins (CD81, CD63, HSP70, and Calnexin) using Western blot assay. The particle size analysis was implemented as follows: 10-20 µl of Exos were diluted with PBS to 1 ml, and then placed on the Nanosight LM10 system at a constant flow rate of 25°C. The transmission electron microscopic observation process was as follows: 20 µl of Exos were dropped onto the sample-loaded copper mesh. After standing for 2 min, the samples were colored with 3% (w/v) sodium phosphotungstate (12501-23-4, Sigma, St. Louis, MO) for 5 min. The mesh was rinsed with PBS to remove excess phosphotungstic acid solution before microscopy.
Cellular uptake of BMSCs-Exos
The PKH26 (Sigma Aldrich, Merck KGaA, Darmstadt, Germany) assay kit was employed for detecting the cellular uptake of Exos. Specifically, the Exos were labeled with PKH26 dye, and then harvested after ultracentrifugation at 100,000 × g for 60 min. PBS was added for rinsing and resuspension. Then, the PKH26-labeled Exos were co-cultured with LX-2 cells for 12 h. The cells were fixed with 4% PFA, then stained with DAPI, and observed under a confocal microscope (Olympus, Tokyo, Japan).
Intervention with miR-144-3p inhibitor
Low expression of miR-144-3p was achieved by transfecting miR-144-3p inhibitor (transfection dosage of 100 nM [19]) or inhibitor negative control (NC), which were from Hanbio (Shanghai, China) into BMSCs. After 48 h, the Exos were extracted and isolated for subsequent experiments. All subsequent experiments were conducted 48 h after transfection.
Cell treatment
LX-2 cell activation was induced as previously described [20]. LPS (10 µg/ml) was added to DMEM medium, which was co-cultured with LX-2 cells for 24 h before subsequent experiments.
Cell transfection
LX-2 cells were transfected with sh-SLC7A11 (transfection dosage of 100 nM [19]), sh-NC, SLC7A11 overexpression vector (oe-SLC7A11, transfection dosage of 2 µg, pcDNA3.1 as vector [21]), and oe-NC, all purchased from Hanbio (Shanghai, China). Specifically, the cells were evenly paved on a six-well plate and cultured until about 70% confluence. Then, Lipofectamine 2000 and corresponding plasmids were added at a 2 : 1 ratio to the six-well plate. After 4-6 h, the culture medium was renewed. After 48-h transfection, the cells were incubated with LPS (10 µg/ml) for 24 h for relevant tests.
Detection of iron content
The Iron Assay Kit (ab83366, Abcam, Cambridge, MA) was used to detect relative cellular ferrous ion (Fe2+) levels in cell lysates. After treatment, LX-2 cells were collected and washed three times in cold PBS. Then, 200 µl of iron assay buffer was added to each well on ice. The collected cell lysate was added with iron reducing agent to reduce switching from Fe3+ to Fe2+. After reaction for 30 min, the lysate was mixed thoroughly with 100 µl of iron probe and incubated in a dark environment at 25°C for 60 min. Finally, a microplate reader was used to measure the absorbance at 593 nm.
Detection of MDA and GSH levels
An MDA content detection kit (E-BC-K028-M, Elabscience, Wuhan, China) and a GSH detection kit (S0053, Beyotime, Shanghai, China) were used to assess the changes in MDA content and GSH level.
Detection of ROS levels
The detection of ROS content in cells was conducted according to previous literature [22]. In short, cells were treated with DCFH-DA (Biolab Technology, Beijing, China) at 37°C for 10 min. PBS was added to remove the remaining DCFH-DA solution, followed by DAPI treatment. Fluorescence images were collected under a fluorescence microscope (Olympus IX51, Tokyo, Japan).
Western blot assay
Cells or tissues were lysed on ice with RIPA buffer (Biyuntian, Shanghai, China), and then the protein concentration was measured with a BCA test kit (Thermo Fisher, Waltham, MA). After that, 8-12% SDS-PAGE gel was used to separate the equivalent amount of protein, and then transferred to 0.22-µm PVDF membrane, which was then blocked with 5% skim milk. Antibodies against HSP70 (ab2787), CD63 (ab134045), CD81 (ab79559), calnexin (ab22595), GPX4 (ab125066), SLC7A11 (ab175186), collagen I (ab138492), CTGF (ab209780), α-SMA (ab5694), and GAPDH (ab181602) diluted at 1 : 1000 from Abcam were incubated together with the membrane overnight at 4°C. Then, secondary antibodies (1 : 2000, ab205718 or 1 : 2000, ab205719, Abcam) were added for 1-h incubation. Following color development, detection was performed using a chemiluminescence imaging system (BioRad, Hercules, CA).
RT-qPCR
Total RNA was extracted from cells or Exos using TRIZOL reagents (Invitrogen, Carlsbad, CA), followed by the synthesis of complementary DNA using PrimerScript reverse transcription kits (for miRNA, B532451-0020, Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China) or cDNA reverse transcription kits (for mRNA, RR047A, Takara, Japan). Then, the product was placed in a –80°C refrigerator for PCR reaction to avoid repeated freeze-thaw. RT-qPCR was implemented using SYBR GreenMix (Takara) on the ABI7500 Real Time PCR system (ABI, Foster City, CA). Data analysis was performed using the 2-ΔΔCt method [23]. GAPDH was selected as an internal reference, and the information about primers and PCR is listed in Table 1.
Table 1
Primer sequences
Dual-luciferase reporter gene experiment
Based on the predicted binding sites, reporter gene vectors carrying the wild sequence (wt-SLC7A11) and mutant sequence (mut-SLC7A11) were designed and synthesized. Subsequently, Lipofectamine 2000 was used for co-transfecting the reporter gene vector with the corresponding miR-144-3p mimic, miR-144-3p inhibitor, mimic NC, or inhibitor NC at a transfection dose of 100 nM into LX-2 cells. Forty-eight hours after transfection, the dual-luciferase detection kit (Promega, Madison, WI) was utilized to detect the luciferase activity in the LX-2 cell lysate.
RNA pull down
RNA pull-down experiment was done using a Pierce magnetic RNA protein pull-down kit (Thermo Fisher Scientific). Specifically, the cell lysate was incubated with biotinylated miR-144-3p probes or nanoclusters (Geneseed, Guangzhou, China) at 4°C for 3 h and then with streptavidin Sepharose beads (Sigma-Aldrich) for 2 h. RNA was extracted using the Trizol method, and the expression of SLC7A11 in the washed RNA complex was detected by RT-qPCR.
Statistical analysis
The data were statistically analyzed by GraphPad prism7 software and all expressed as mean ± standard deviation (x ±s). The comparison between the two groups was carried out using the t-test, and that among multiple groups was conducted using one-way ANOVA. Post-hoc multiple comparisons were performed using Tukey’s multiple comparisons test. Statistical significance was set at p < 0.05.
Results
Isolation and identification of BMSC-Exos
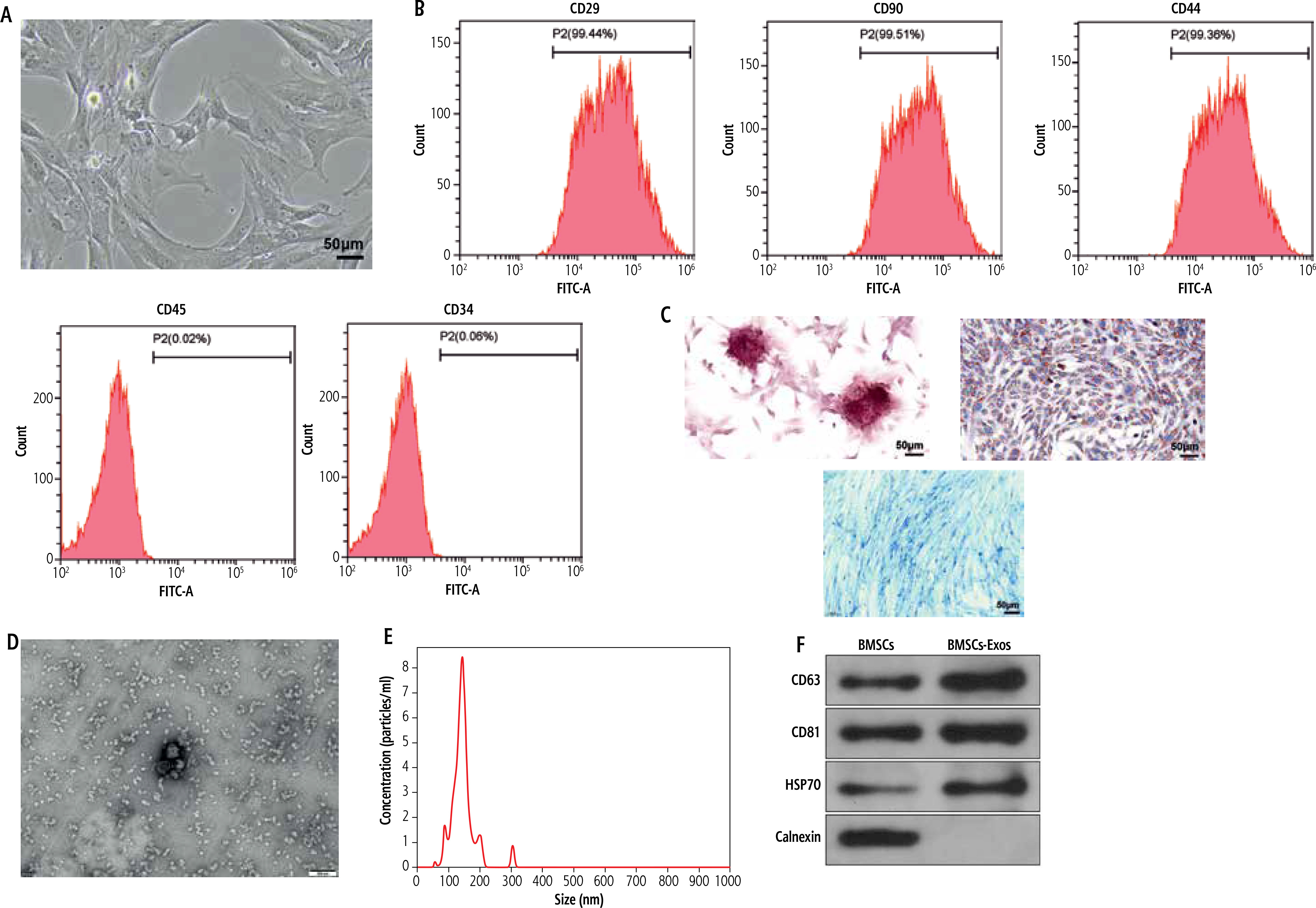
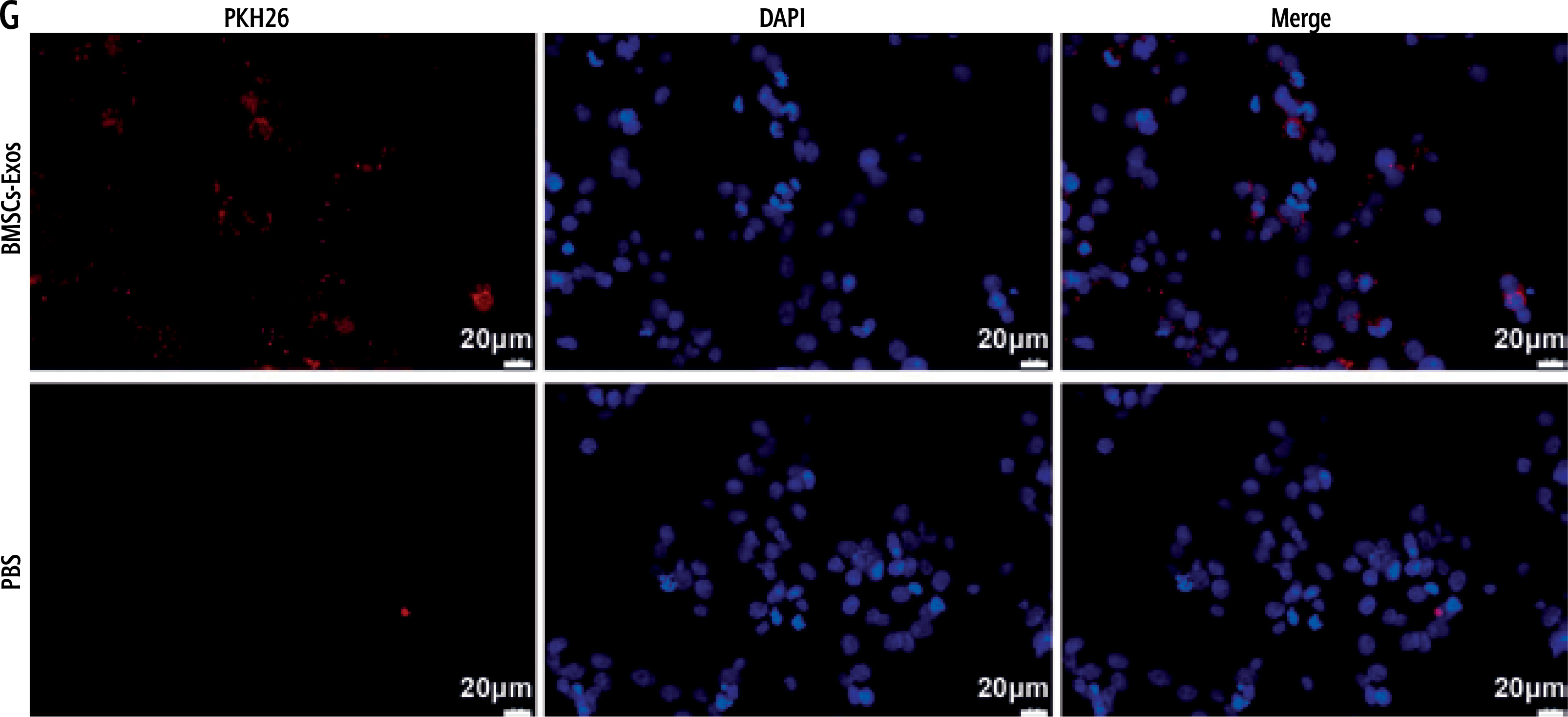
Mesenchymal stem cells (MSCs) have become the most promising candidate cells for the treatment of liver fibrosis due to their continuous self-renewal, multi-directional differentiation potential, low immunogenicity, and minimal invasiveness. To clarify the impact of BMSCs on HSC activation, we first identified the extracted BMSCs. Under the microscope, BMSCs exhibit a typical vortex-like arrangement (Fig. 1A). The surface markers of BMSCs detected by flow cytometry showed high expression of CD29, CD90, and CD44, but no expression of CD45 and CD34 (Fig. 1B). The three-lineage differentiation identification results showed that BMSCs were all positive (Fig. 1C). Subsequently, we identified the extracted BMSC-Exos and observed their morphology under a transmission electron microscope. BMSC-Exos were spherical- or elliptical-shaped, with uniform size (Fig. 1D); nanoparticle tracking analysis (NTA) revealed that the particle size of BMSC-Exos was within the range of 80-160 nm (Fig. 1E). Western blot analysis showed that compared with BMSCs, CD81, CD63, and HSP70 were significantly overexpressed but calnexin was not expressed in BMSC-Exos (Fig. 1F). Eventually, we detected the uptake of PKH26-tagged Exos by LX-2 cells. After 12 h of co-incubation, a large amount of Exos were internalized by LX-2 cells and completely distributed around the nucleus, while no PKH26-labeled fluorescence was observed in the PBS group (Fig. 1G).
Fig. 1
Isolation and identification of BMSC-Exos. A) Morphology of BMSCs under the microscope; B) Identification of expression of surface antigen markers in BMSCs by flow cytometry; C) Identification of osteogenic differentiation, adipogenic differentiation, and chondrogenic differentiation of BMSCs by Alizarin red staining, oil red O staining, and Alcian Blue staining; D) Morphology of BMSC-Exos observed under TEM; E) NTA particle size analysis of BMSC-Exos; F) Western blot detection of expression of CD81, CD63, HSP70, and calnexin G) Immunofluorescence observation of uptake of BMSC-Exos by LX-2 cells; the experiment was repeated 3 times
BMSC-Exos can affect activation of HSCs by regulating ferroptosis
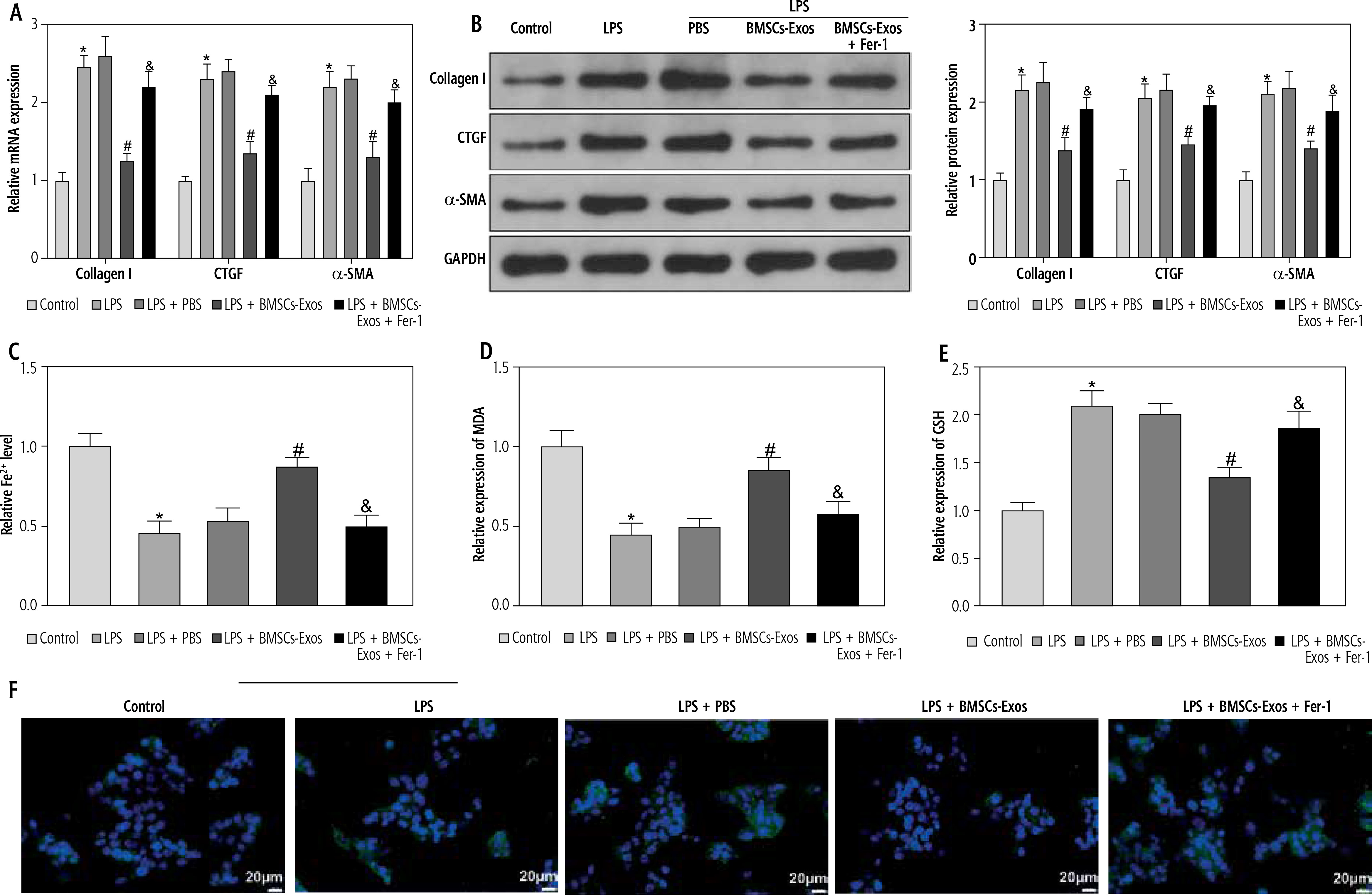
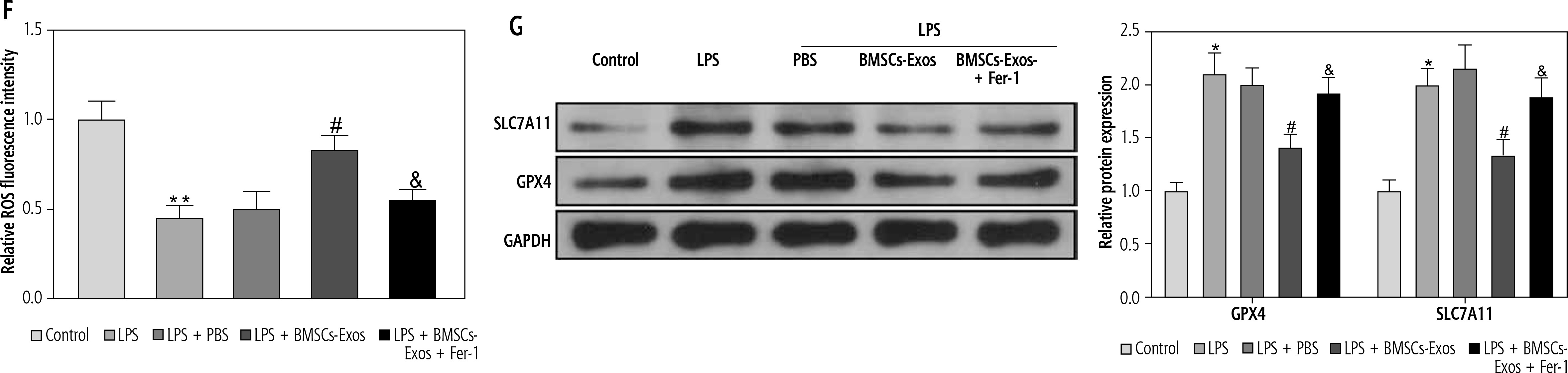
Subsequently, we observed the effect of BMSC-Exos on LPS-evoked activation of HSCs, and detected the expression of fibrosis markers (collagen I, CTGF, and α-SMA). LPS markedly increased the expression of fibrosis markers (Fig. 2A, B). However, it should be noted that BMSC-Exos remarkably suppressed the expression of fibrosis markers (Fig. 2A, B). Given that Exos can inhibit HSC activation by facilitating Exos processes [24], we speculated that BMSC-Exos might affect the activation process of HSCs by promoting ferroptosis in LX-2 cells. For this reason, we observed the changes related to ferroptosis in LX-2 cells after BMSC-Exos treatment. The results showed that the LPS caused a significant decrease in cellular iron content, MDA, and ROS levels but a significant reduction in the GSH level (Fig. 2C-F). The Western blot results showed a significant increase in the expression of ferroptosis-associated proteins GPX4 and SLC7A11 in the LPS group versus the control group (Fig. 2G). Intervention with BMSC-Exos notably counteracted this effect of LPS (Fig. 2C-G). Interestingly, when LX-2 cells were treated with both BMSC-Exos and ferroptosis inhibitor Fer-1 (Sigma, USA, 0.1 µM) [25, 26], the aforementioned observed effects of BMSC-Exos on HSC activation and ferroptosis were significantly reversed by Fer-1 (Fig. 2A-G). These results indicated that BMSC-Exos affected the activation of HSCs by regulating ferroptosis.
Fig. 2
BMSC-Exos mediates HSC activation by regulating ferroptosis. A, B) RT-qPCR and Western blot detection of expression of fibrosis markers (collagen I, CTGF, and α-SMA); C-F) Iron content as well as levels of MDA, GSH, and ROS detected by corresponding reagents and DCHA-DA molecular probe F) Iron content as well as levels of ROS detected by corresponding reagents and DCHA-DA molecular probe; G) Western blot detection of expression of ferroptosis-associated proteins GPX4 and SLC7A11. Data are expressed as mean ± standard deviation; the cell experiment was repeated 3 times. *p < 0.05, **p < 0.01 vs. Control group, #p < 0.05 vs. LPS + PBS group, &p < 0.05 vs. LPS + BMSC-Exos group
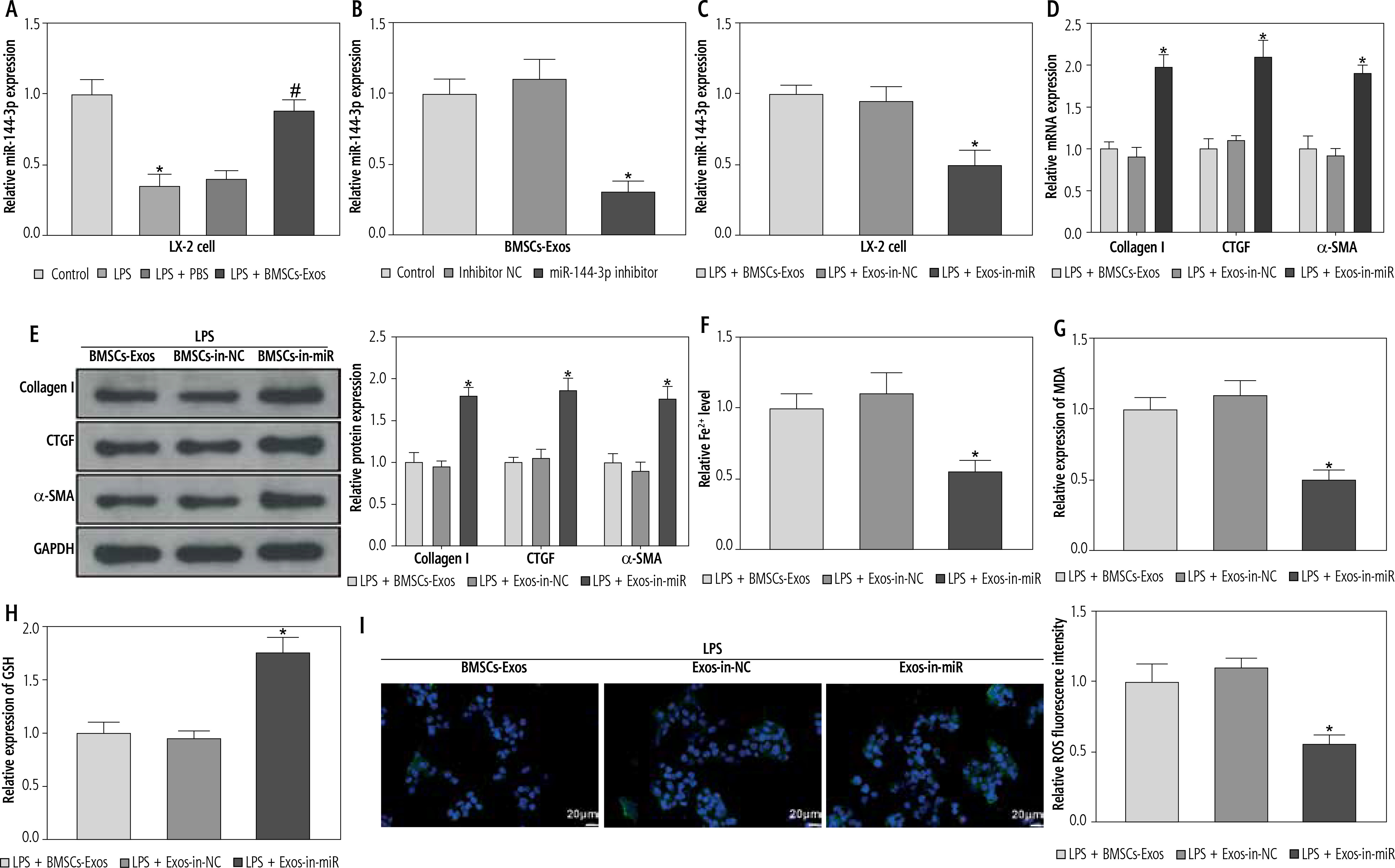
miR-144-3p from BMSC-Exos regulates HSC activation by mediating ferroptosis
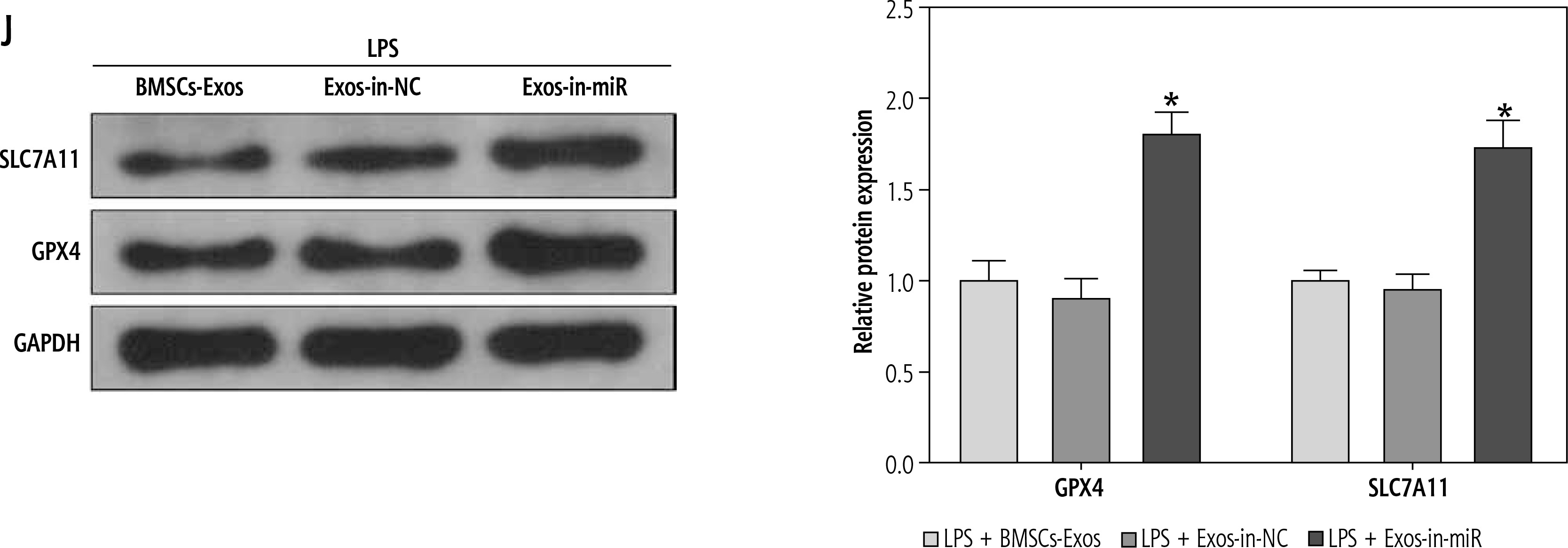
In the above experiment, we observed that BMSC-Exos affected the activation of HSCs by regulating ferroptosis, but the relevant molecular mechanism remained unclear. Several studies have suggested that miR-144-3p could play a significant role in modulating renal injury or myocardial ischemia-reperfusion by regulating ferroptosis [14, 15], but its function in liver fibrosis is scarcely explored. Subsequently, we analyzed the expression of miR-144-3p in cells after different treatments using RT-qPCR. The results showed that relative to the control group, miR-144-3p expression was significantly reduced in the LPS group; intervention with BMSC-Exos markedly elevated miR-144-3p expression (Fig. 3A). To further substantiate our hypothesis, we transfected miR-144-3p inhibitor and corresponding inhibitor NC into BMSCs and extracted BMSC-Exos subsequently. RTq-PCR detection was performed to examine transfection efficiency, and we found that miR-144-3p inhibitor led to a significant reduction in miR-144-3p expression in BMSC-Exos (Fig. 3B). Afterwards, we co-incubated the Exos from miR-144-3p inhibitor-transfected BMSCs (named Exos-in-miR) with LPS-treated LX-2 cells. The RTq-PCR data showed that miR-144-3p expression in the Exos-in-miR group was remarkably reduced as compared to the Exos-in-NC group (Fig. 3C). Furthermore, we evaluated relevant cellular protein changes and found that Exos-in-miR treatment up-regulated the expression of liver fibrosis markers as well as GSH, GPX4, and SLC7A11 while reducing iron content and MDA and ROS levels in LX-2 cells (Fig. 3D-J). To conclude, miR-144-3p originating from BMSC-Exos affected ferroptosis to mediate HSC activation.
Fig. 3
miR-144-3p from BMSC-Exos affects ferroptosis to regulate HSC activation. A-C) RT-qPCR determination of expression of miR-149-5p in LX-2 cells and Exos, respectively; D, E) RT-qPCR and Western blot detection of expression of fibrosis markers (collagen I, CTGF, and α-SMA); F-I) Iron content as well as levels of MDA, GSH, and ROS J) Western blot detection of expression of ferroptosis-associated proteins GPX4 and SLC7A11; data are expressed as mean ± standard deviation; the cell experiment was repeated 3 times. *p < 0.05 vs. Control, inhibitor NC or LPS + Exos-in-NC group, #p < 0.05 vs. LPS + PBS group
miR-144-3p from BMSC-Exos negatively mediates SLC7A11 expression
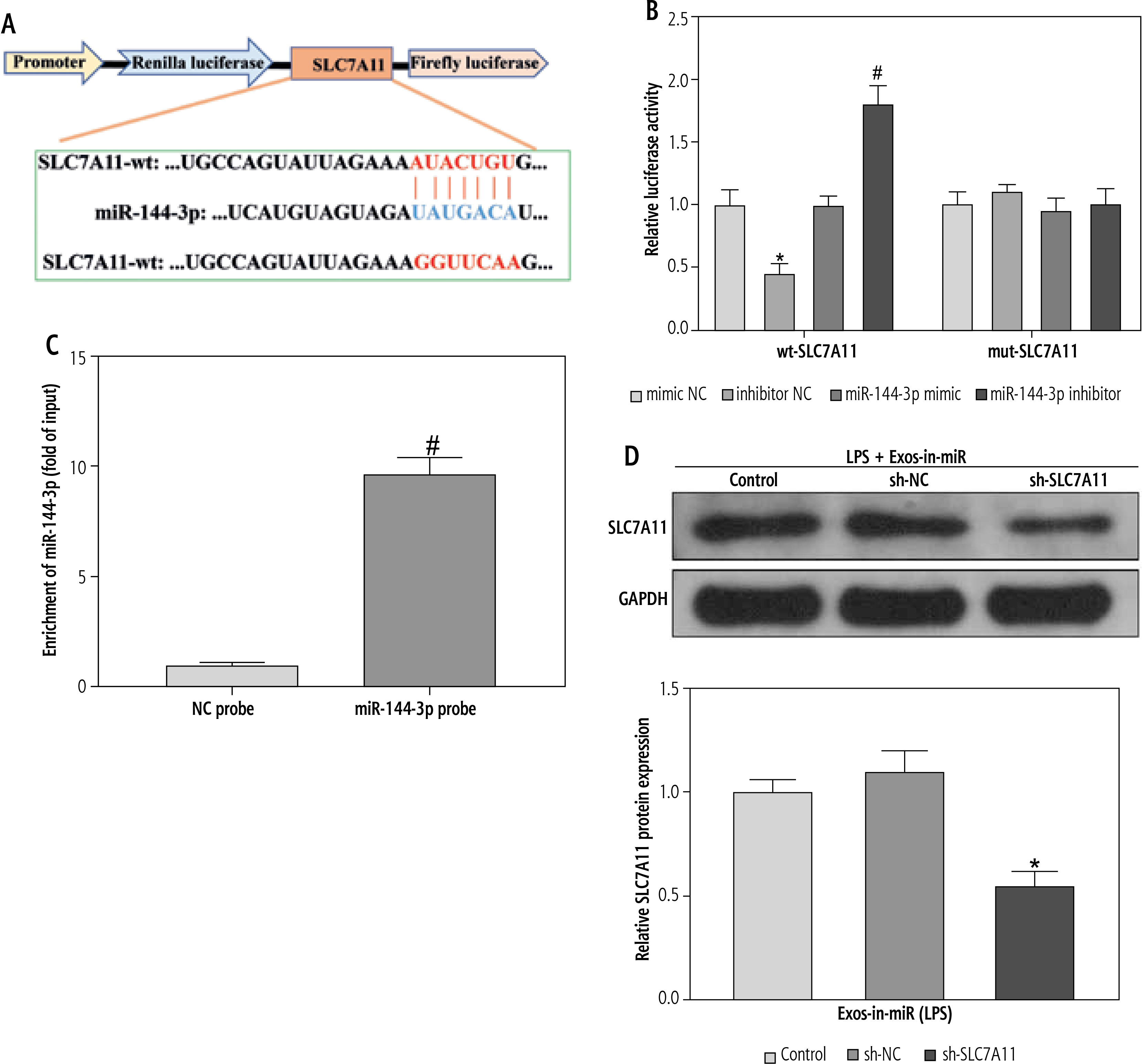
The next focus was to unravel the downstream molecular mechanism by which miR-144-3p from BMSC-Exos regulated ferroptosis and HSC activation. The TargetScan database predicted that SLC7A11 might bind to miR-144-3p as a downstream target gene (Fig. 4A). To further verify the binding of miR-144-3p to SLC7A11, we conducted a dual-luciferase reporter gene experiment. It was observed that miR-144-3p mimic sharply down-regulated the luciferase activity of wild-type wt-SLC7A11, while miR-144-3p inhibitor resulted in an opposite effect (Fig. 4B). The RNA pull-down experiment further evidenced that SLC7A11 could be captured by biotinylated miR-144-3p probes (Fig. 4C), indicating SLC7A11 to be a potential target for miR-144-3p. To further investigate their relationship, Exo-in-miR was co-incubated with LPS-stimulated LX-2 cells transfected with sh-SLC7A11. Consequently, the SLC7A11 expression was significantly reduced (Fig. 4D). The aforesaid results demonstrated that miR-144-3p originating from BMSC-Exos targeted and negatively modulated the expression of SLC7A11.
Fig. 4
Exosomal miR-144-3p targets SLC7A11. A) Binding sites and mutation sequences of miR-144-3p and SLC7A11; B, C) Relationship between miR-144-3p and SLC7A11 validated by dual-luciferase reporter gene experiment and RNA pull-down experiment; D) Western blot detection of expression changes of SLC7A11; data are expressed as mean ± standard deviation; the cell experiment was repeated 3 times. *p < 0.05 vs. mimic NC or LPS + Exos-inmiR + sh-NC group, #p < 0.05 vs. inhibitor NC or NC probe group
miR-144-3p from BMSC-Exos regulates ferroptosis to affect HSC activation through down-regulating SLC7A11
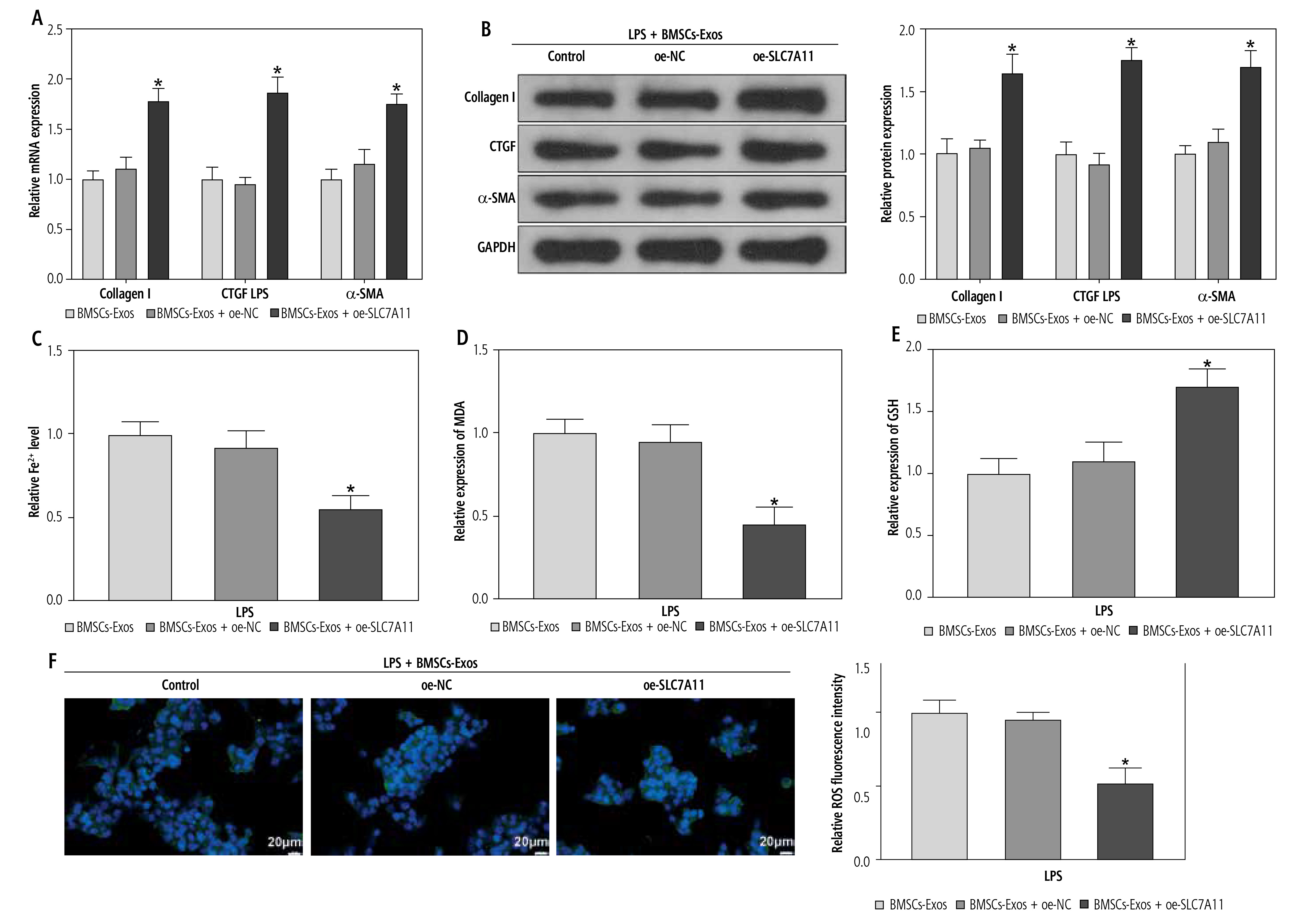
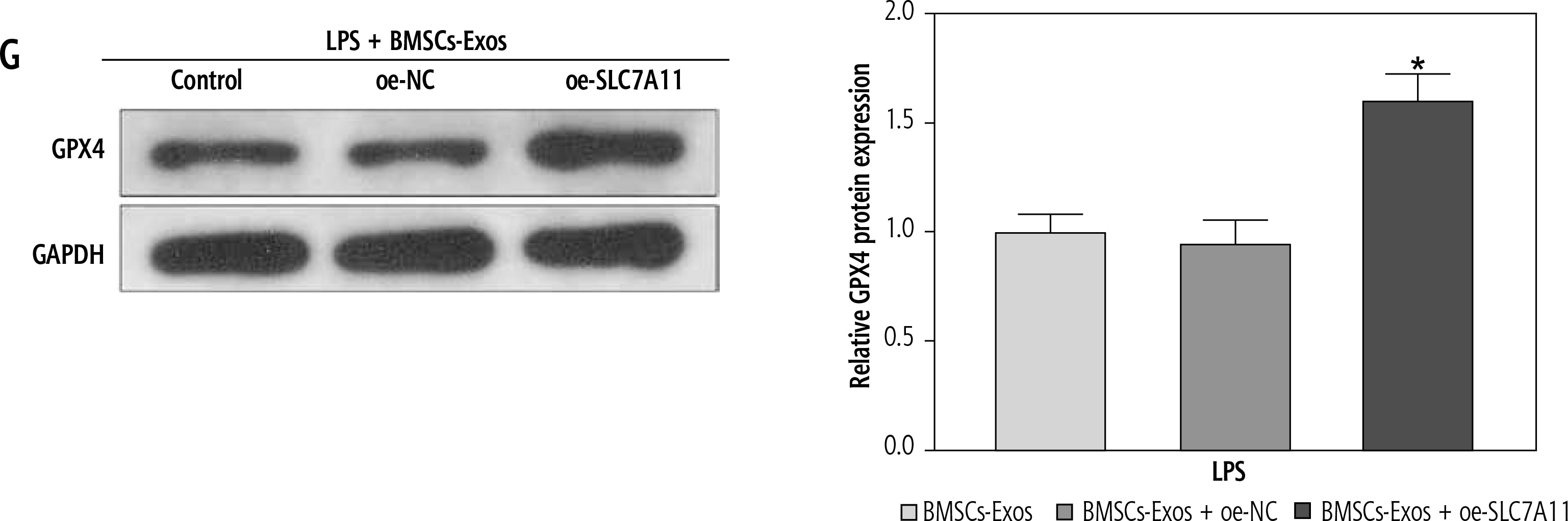
Ultimately, to clarify whether miR-144-3p, sourced from BMSC-Exos, targeted SLC7A11 to regulate ferroptosis and affect HSC activation, we investigated the impacts of BMSC-Exos combined with oe-SLC7A11 on ferroptosis and activation of HSCs. Intervention with oe-SLC7A11 counteracted the effect of BMSC-Exos alone on ferroptosis and activation of HSCs, sharply inhibiting ferroptosis and promoting HSC activation (Fig. 5A-G). Conclusively, miR-144-3p originating from BMSC-Exos negatively regulated the expression of SLC7A11, thereby accelerating ferroptosis and affecting HSC activation.
Fig. 5
miR-144-3p from BMSC-Exos regulates ferroptosis to affect HSC activation through down-regulating SLC7A11. A, B) RT-qPCR and Western blot detection of fibrosis markers (collagen I, CTGF, and α-SMA); C-F) Detection of iron content, MDA, GSH, and ROS levels G) Western blot detection of the expression of ferroptosis-related protein GPX4; data are expressed as mean ± standard deviation; the cell experiment was repeated 3 times. *p < 0.05 vs. LPS + BMSCs-Exos + oe-NC group
Discussion
Hepatic stellate cells perform critical functions in acute liver damage of diverse etiologies and act as the primary fibrogenic cells in chronic liver damage [27]. Transdifferentiation (or “activation”) of HSCs, the primary origin of myofibroblasts, functions as the main factor that can drive liver fibrogenesis [28]. MSCs exert direct anti-fibrotic effects on HSCs, repressing cell proliferation and ECM production in HSCs and stimulating their apoptosis [29]. In the present study, we found that BMSC-Exos could boost ferroptosis to suppress HSC activation, which was related to miR-144-3p-mediated inhibition of SLC7A11.
The first finding of our experiments was that BMSC-Exos could block the activation of HSCs, corresponding to markedly down-regulated levels of fibrosis markers (collagen I, CTGF, and α-SMA). A diverse range of cell types in the liver such as hepatocytes and liver sinusoidal endothelial cells can communicate with HSCs via Exos, which in turn mediate the biological functions of HSCs [30]. More importantly, MSC-isolated Exos have shown great potential to affect HSC activation in the context of liver fibrosis. For instance, Exos sourced from human adipose MSCs, namely hADMSCs-Exos, relieve HSC activation and delay liver fibrogenesis, showing promise as extracellular nanovesicle-based therapeutics for patients with liver fibrosis [31]. MSC-Exos can repress HSC activation, showing anti-fibrotic potential in the liver [32]. Rong et al. administered human BMSC-Exos into CCl4-treated rats and found that BMSC-Exos effectively attenuated liver fibrosis via suppression of HSC activation, which was achieved through blockade of the Wnt/β-catenin pathway [10]. Also, BMSC-Exos restrained the activation of HSCs through regulation of miR-192-5p/PPP2R3A [33]. Another study also revealed that human umbilical cord MSC-derived Exos (hUC-MSC-Exos) repressed HSC activation via the miR-148a-5p/SLIT3 axis and thus attenuated the progression of liver fibrosis [34]. Li et al. reported a similar finding that hUC-MSC-Exos could protect against liver fibrosis through HSC activation [35], and their new study further demonstrated that MSC-Exos stimulated the ferroptosis of HSCs [24]. Our experiments suggested that BMSC-Exos reversed the reductions in iron content, MDA, and ROS as well as elevations in the levels of GSH and ferroptosis-relevant proteins GPX4 and SLC7A11 stimulated by LPS, indicating that BMSC-Exos enhanced the ferroptosis of HSCs. Fer-1, an inhibitor of ferroptosis, was found in this study to reverse the impact of BMSC-Exos on HSC activation and ferroptosis. It is rational to conclude that BMSC-Exos suppressed HSC activation by promoting ferroptosis.
We next focused on the mechanism by which BMSC-Exos affected HSC activation and ferroptosis. miR-144-3p expression was determined to be up-regulated in HSCs upon co-incubation with BMSC-Exos. A pro-ferroptotic effect of miR-144-3p has been reported in osteosarcoma [36]. Also, a recent study demonstrated that miR-144-3p can accelerate high glucose-caused ferroptosis by targeting USP22 [37]. Although there is no study showing the role of miR-144-3p in HSC activation and even liver fibrosis, miR-144-3p was observed to affect cardiac fibrosis following myocardial infarction [38]. SLC7A11 has been proposed to be a target of miR-144-3p in several studies, and their relationship plays a role in the progression of diseases such as endometrial cancer and ulcerative colitis [13, 39]. The study of Jian et al. reported that Ginsenoside Re ameliorated myocardial ischemia/reperfusion-related ferroptosis through miR-144-3p-mediated SLC7A11 [15]. Moreover, ulinastatin may inhibit miR-144-3p to attenuate LPS-evoked podocyte ferroptosis by rescuing SLC7A11 expression [14]. Ferroptosis, iron-dependent cell death triggered by lipid peroxidation, is modulated by complicated oxidation and antioxidant systems. As a key inhibitor of ferroptosis, the activity of GPX4 relies on glutathione released upon activation of the cystine-glutamate antiporter SLC7A11 [40]. Combined with the results in this study indicating that miR-144-3p targeted and down-regulated SLC7A11, it was reasonable to speculate that miR-144-3p might promote ferroptosis by targeting SLC7A11. Not surprisingly, SLC7A11 overexpression counteracted the regulatory effects of BMSCs-Exos on the activation and ferroptosis of HSCs. Therefore, BMSCs-Exos might deliver miR-144-3p to reduce SLC7A11 expression, thereby facilitating the ferroptosis and blocking the activation of HSCs.
In conclusion, our study suggested that the transfer of miR-144-3p via BMSCs-Exos contributes to blocking the activation of HSCs through its pro-ferroptotic function. This finding provides a deeper understanding of the mechanisms relevant to HSC activation in liver fibrosis and also lays a foundation for developing new anti-fibrotic targets. However, more attention should be paid to data validation in animal experiments and clinical trials, considering the unsatisfactory extraction and culture of pure Exos at present.