






Biotin is a crucial vitamin that dissolves in water. It serves as a cofactor for four carboxylase enzymes in the human body: propionyl Co-A carboxylase, acetyl Co-A carboxylase, pyruvate carboxylase, and 3-methylcrotonyl Co-A carboxylase. Propionyl Co-A carboxylase and methylcrotonyl Co-A carboxylase are enzymes that play a role in the breakdown of leucine, isoleucine, and valine [1].
Biotinidase (BTD) enzymatically transforms the precursor molecule biocytin into biotin. Biotin is a crucial coenzyme for carboxylase enzymes, which play a vital role in the synthesis of fatty acids, gluconeogenesis, and the breakdown of branched-chain amino acids. Biotinidase deficiency (BD) is an autosomal recessive metabolic condition. This condition is caused by specific changes in the BTD gene, which lead to a decrease or absence of biotinidase activity, and it is a classic example of vitamin-responsive disease [2]. People with BD lack the ability to metabolize biotin, leading to a secondary shortage of this nutrient. Biotinidase deficiency occurs as a result of mutations in the biotinidase gene. The BTD gene is located in the 3q25.1 locus, consisting of four exons and encoding 543 amino acids. Researchers have identified more than 150 different mutations in the BTD gene to date [3]. The incidence of the disease is 1 : 60 000 around the world, while the incidence in Turkey is higher with a 1 : 11 000 ratio [4].
Biotinidase deficiency is classified into two groups: significant and moderate deficiency, based on enzyme activity levels. A severe biotinidase deficit is defined by blood enzyme activity below 10%, while a partial BD is defined by serum enzyme activity between 10% and 30% [5].
The age at which symptoms appear can vary, spanning from a few weeks after birth to 8 years of age, depending on whether the BD is partial or total. The earliest clinical manifestations of BD exhibit considerable variability; nevertheless, the majority of affected persons experience neurological and cutaneous symptoms in childhood. These symptoms may include, but are not limited to, seizures, muscle weakness, lack of coordination, hearing impairment, delayed development, hair loss, and skin rash. The symptoms of the central nervous system (CNS) occur as a result of the buildup of biocytin, biotinyl peptides, and lactate in the cerebrospinal fluid. Children who have a partial deficiency of the biotinidase enzyme may not show any symptoms or may experience only persistent seborrheic dermatitis that is difficult to treat [6].
The clinical symptoms mostly affect the skin and central nervous system. The child typically exhibits symptoms such as hair loss, seizures, weak muscle tone, delayed development, fungal infection, lack of coordination, damage to the optic nerve, hearing loss, and inflammation of the skin. T cell malfunction can also manifest, potentially causing immunodeficiency and subsequent susceptibility to opportunistic infections. Respiratory distress may manifest as hyperventilation, laryngeal stridor, or apnoea [7]. Cutaneous signs encompass skin rash, dermatitis, alopecia, and conjunctivitis, as well as viral and fungal infections resulting from immunological dysfunction. Scaly red patches on the inner surfaces of the joints and around the mouth, as well as outbreaks resembling seborrheic dermatitis, might be observed. Severe instances may include crusting, lichenification, and open lesions that have the potential to develop secondary infections. Disruptions in lipid metabolism and changes in the skin’s composition may contribute to the development of skin symptoms in individuals with BD [8]. Thankfully, regularly giving children biotin in pill form can effectively alleviate a majority of symptoms and prevent the onset of symptoms in children who have not yet shown any signs, if started early. Administering a daily dose of 5–20 mg of biotin as a supplement results in significant clinical and biochemical enhancements [9]. There is a suggestion that administering lesser amounts of biotin, specifically 5 to 10 mg/day, may be adequate for treating children with incomplete BD [10]. Infants that are diagnosed with severe biotin deficiency (BD) early in the neonatal period and get biotin therapy with high adherence may experience normal physical and cognitive growth [11]. In cases of extensive BTD, certain symptoms may remain permanent despite the use of biotin therapy. The management of patients with BD should encompass a comprehensive approach involving an annual ophthalmologic examination, auditory testing, and evaluation by a metabolic specialist.
Radioassay can quantify biotinidase activity in serum, plasma, fibroblasts, leukocytes, and other tissue extracts. The colorimetric assay is the most commonly employed approach for diagnosing BTD as it allows for the assessment of biotinidase activity in plasma or serum [12, 13]. The typical range of serum biotinidase activity in humans is between 4.4 and 10 nmol/min/ml, with an average activity of 7.1± 1.2 nmol/min/ml [14]. The serum biotinidase activity of carriers may exhibit similarities to those with partial BD, which might complicate the diagnosis when relying solely on enzyme analysis.
Mutation analysis for biotinidase deficiency is valuable for definitive testing to distinguish between patients with severe and moderate biotinidase deficiency, as well as carriers. It is advisable to do biotinidase activity testing on both asymptomatic and symptomatic siblings of patients who are affected [15].
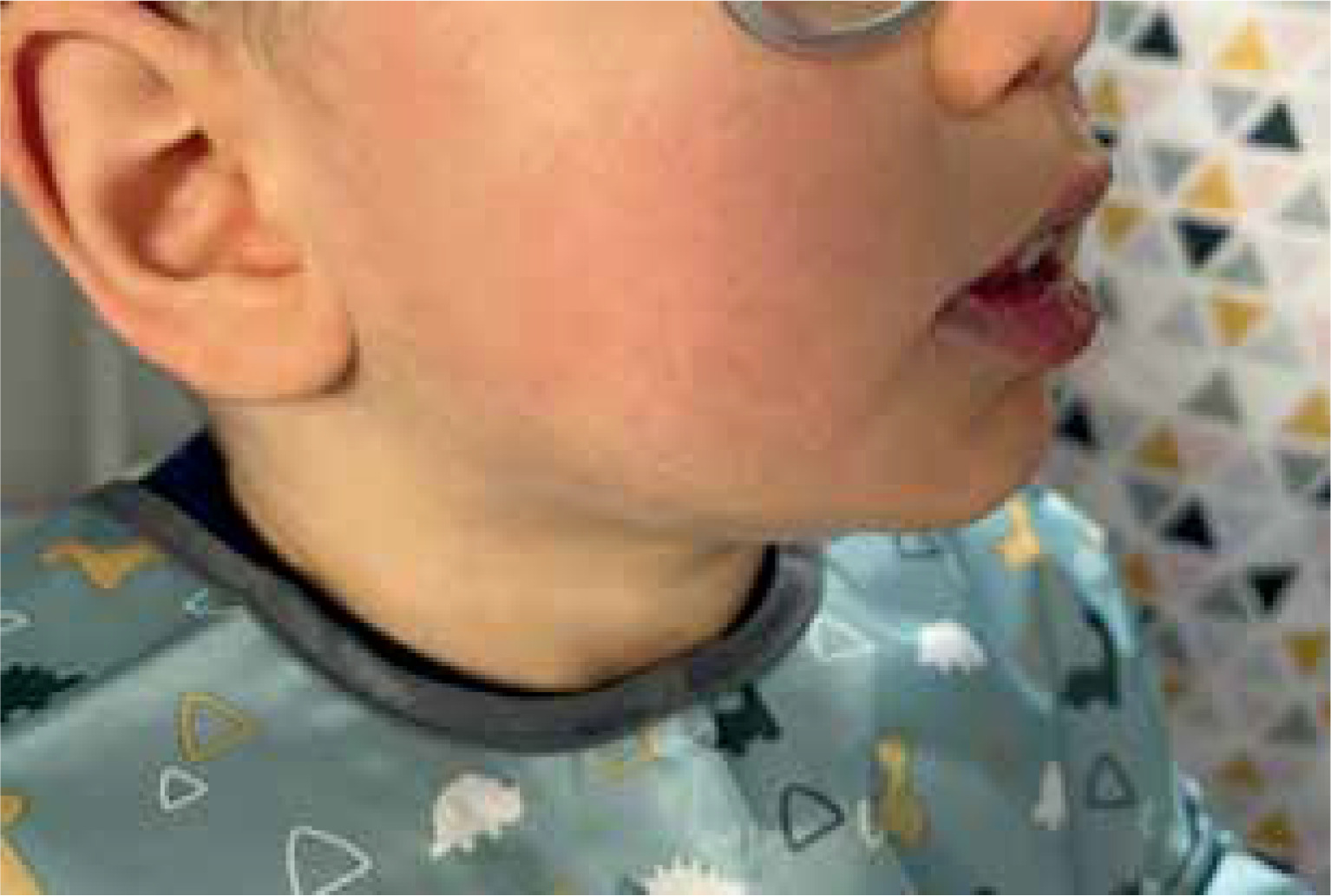
We present the case report of a 16-month-old male with progressive weakening, lethargy, hypotonia, feeding difficulties, developmental delay, and eczematous facial, upper, lower, and trunk rash. Skin abnormalities included redness, scaling, and itching. The child had a history of recurrent skin infections. Clinical examination revealed periorificial and acral dermatitis, keratosis pilaris, and diffuse erythematous patches on the skin (Figures 1 and 2). The brain MRI revealed incomplete myelination, expansion of the subarachnoid space, and a congenital venous abnormality in the right cerebral peduncle. EEG was within typical ranges.
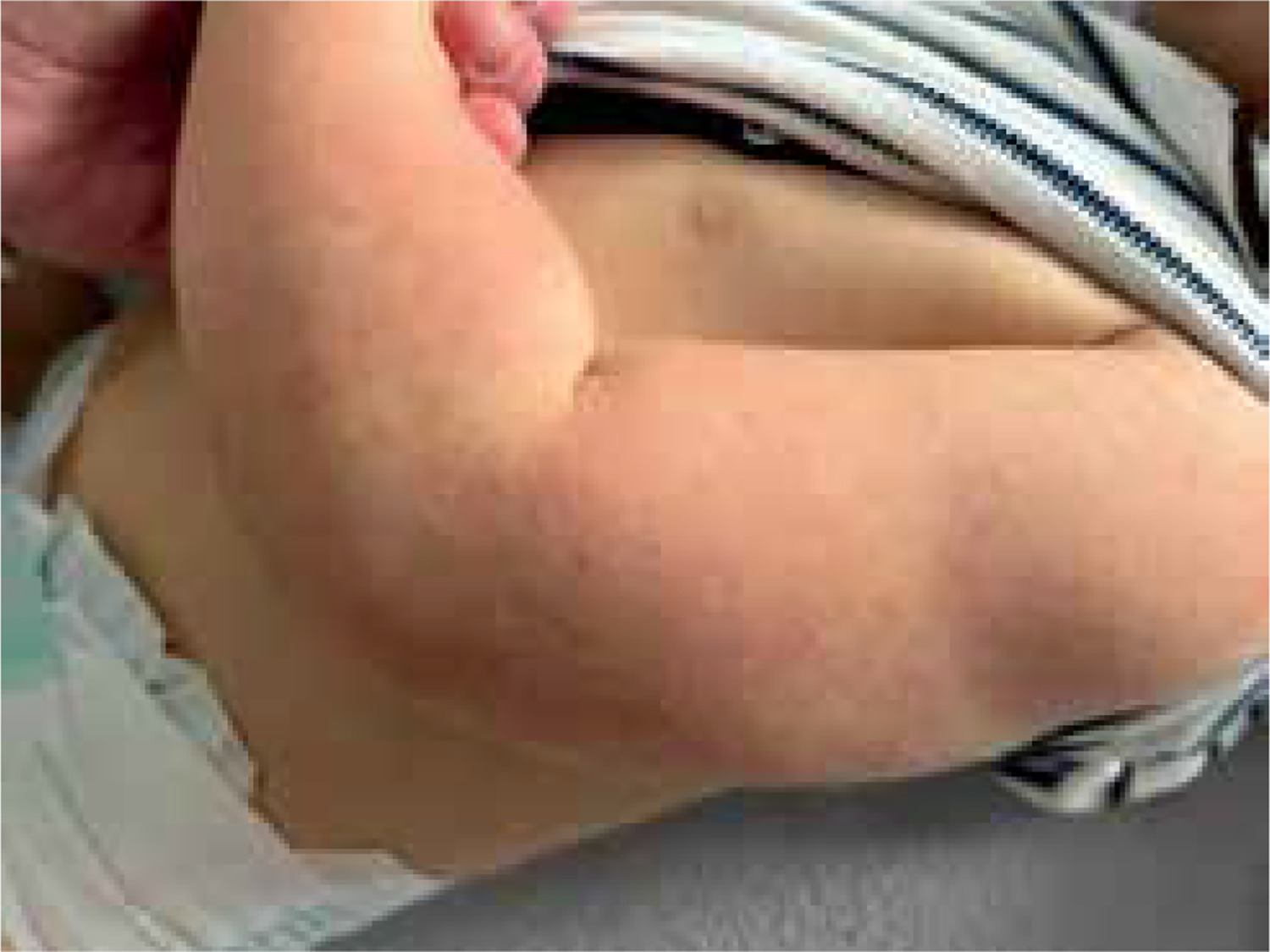
Figure 1
Keratosis pilaris characterized by grouped, horny, keratotic, follicular papules, located over the posterolateral upper arms
The levels of the biotinidase enzyme were significantly low, and enzyme activity was found to be 20%. Genetic testing confirmed the presence of biotinidase deficiency with a heterozygous mutation in the BTD gene. The BTD gene (NM_001281723.c) was shown to have a pathogenic mutation, specifically c.1336G>C (p. Asp446His), in exon 4. This variant was discovered using whole-exome sequencing and was found to be in the heterozygous state. The child had a daily regimen of 10 mg biotin tablets initiated and showed improvement during the three-month follow-up period.