









Introduction
Familial Mediterranean fever (FMF) is an autosomal recessive disease with an autoinflammatory nature. It affects mainly Turkish, Armenian, Arab, and Jewish people [1]. Clinical presentation includes recurrent attacks of fever, polyserositis, and rash [2]. The most critical complication of FMF is amyloidosis, which is responsible for the long-standing morbidity and mortality [3]. Unfortunately, there are no pathognomonic signs for the diagnosis of FMF. Variability in the clinical presentation of FMF makes the diagnosis uncertain, so MEFV genotype remains the only reliable diagnostic tool [4].
FMF is caused by different mutations (more than 300 mutations) that encode a protein – pyrin/marenostrin. The main function of pyrin is its anti-inflammatory role via the autophagy of regulators of innate immunity. The resulting mutant form of pyrin leads to inappropriate activation of neutrophils and uncontrolled production of interleukin-1 (IL-1), leading to episodes of inflammation mainly in the peritoneum, pleura, and joints [5]. The clinical presentation and the development of complications such as amyloidosis are influenced by the type of MEFV mutations [6].
Homozygotes for this mutation are prone to more severe clinical manifestations and amyloidosis [7]. However, the absence MEFV mutations does not rule out a diagnosis of FMF [8].
Although there have been many studies on genetic mutations of FMF, data on the Egyptian population remain very limited.
Aim
We aimed to study the clinical and genetic characterization of familial Mediterranean fever among a cohort of Egyptian patients in the Kafrelsheikh governate.
Material and methods
This is prospective study that was carried out at Kafrelsheikh University Hospital Outpatient Clinic between March 2019 and March 2020. We included all patients who came to our outpatient clinic with symptoms suggestive of FMF (recurrent attacks of abdominal pain and fever), and diagnosis of FMF was confirmed by gene study, and we excluded any FMF patient who had been previously diagnosed and started the treatment and those who refuse to participate or to continue the study.
Participants
One hundred and nine patients were included; however, 9 patients refused to participate in the study, so the final analysis was done for 100 patients only. All patients were subjected to detailed medical history including the number and duration of attacks per year and previous surgical history. We started the treatment with colchicine, and we followed up the patients for confirmation of good control of symptoms and attacks. Complete clinical examination for any palpable organ or scars from operations.
Initial evaluation
Blood samples were tested for amyloid A and the most common 15 MEFV mutations in the Mediterranean region, collected from all participants in EDTA tubes, and DNA was extracted from white blood cells using the FMF Multiplex Real Time PCR Kit (SNP-Biotechnology, Ankara, Turkey). PCR amplification was then carried out according to the manufacturer’s instructions. Exons 1, 2, 3, 5, and 10 were amplified by a PCR program set as follows: 95°C denaturation for 30 s, 55°C hybridization for 30 s, and 72°C elongation for 30 s.
Classification after genetic study
Compound heterozygote: The presence of 2 different mutant alleles at a particular gene locus, one on each chromosome of a pair.
Heterozygous: refers to having inherited different forms of a particular gene from each parent.
Homozygous genotype: where an individual inherits identical forms of a particular gene from each parent.
Follow-up of the patients
The follow-up was done every 3 months for 1 year, recording clinical symptoms and the number and duration of attacks. We confirmed whether the starting dose of 1.5 mg was adequate. The dose was increased gradually from 1.5 to more than 3 mg (0.5 mg every month if the attacks were not controlled or serum amyloid increased).
Abdominal ultrasound examination
Patients also underwent abdominal ultrasound examination for measurement of the spleen longitudinal diameter and for exclusion of any associated disease or complication.
Ethics and consent
This study was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, and applicable regulatory requirements. Written informed consent was obtained from all patients after explanation of the research idea.
Ethical Committee at Kafrelsheikh University, Faculty of Medicine approved the research protocol before starting. IRB 00012367-20-10-006.
Statistical analysis
Data were collected and entered into Microsoft Excel spreadsheets before being transferred to the Statistical Package for the Social Sciences (SPSS) software (SPSS Inc., Chicago, IL, USA) version 16 for Windows 7 (Microsoft Corp., Redmond, WA) to be analysed.
Wilcoxon’s test and paired Student’s t-test were used to compare paired data, whereas the χ2 test, Fisher’s exact test, and the Mann-Whitney U-test were used to compare unpaired data.
Results
Patient demographics
This study included 109 patients confirmed to have FMF by gene mutation. However, 9 patients refused to participate in the study, so final analysis was done on 100 patients, 67 of whom were female and 33 male. The ages of studied patients ranged from 18 to 60 years, with mean 26.21 ±11.84 years. Parental consanguinity was found in 39% while positive family history was seen in 48%; history of abdominal surgery was found in 57% (Table I).
Clinical presentation of FMF patients
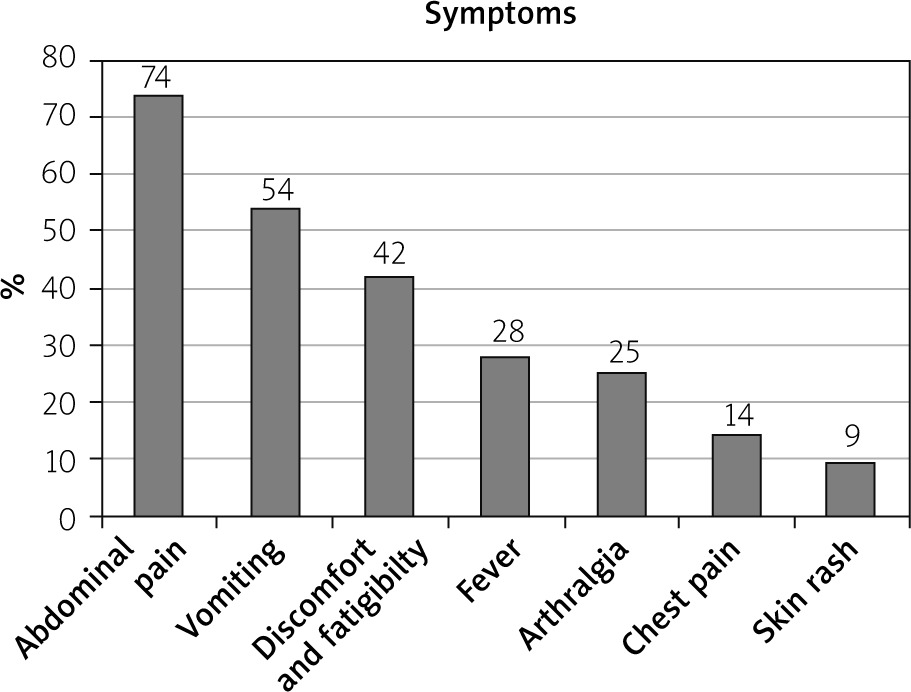
Although FMF is classified as a fever, fever was present only in 28%; abdominal pain was among the most common presentation, seen in 74%, and it was associated with vomiting (54%) followed by arthralgia (25%), and chest pain and skin rashes (9%) (Figure 1).
Sonographic findings recorded in FMF patients
The range of splenic size was 10.9–17 cm and the mean splenic size for the studied patients was 12.17 ±1.35 cm. Minimal pelvic collection (during an attack) was recorded in 6% only (Table II).
FMF genetics and relation to attacks
Table III show different genetic mutations of the studied patients; the most common one is E148 Q, seen in 31%. There is a statistically significant difference between the number of attacks per year and the serum amyloid level in patients with homozygote, heterozygote, or compound mutations, with p-value < 0.001, 0.007 respectively, whereas no difference in the attack duration, p-value 0.287 (Table IV).
Table III
Different genetic mutations of the studied patients (N = 100)
Table IV
Relationship between mutation type and the attacks
Response to treatment
Table V shows that response to a colchicine dose less than 1.5 mg/day was seen in 11.76% of heterozygote patients, in 26.67% of compound heterozygote patients, and in 23.53% of homozygote patients, with a p-value of 0.011.
Table V
Response to different doses of colchicine in FMF patients
| Colchicine dose response [mg/day] | Mutation status | ||||||
|---|---|---|---|---|---|---|---|
| Heterozygous (n = 68) | Compound heterozygote (n = 15) | Homozygous (n = 17) | P-value | ||||
| < 1.5 | 8 | 11.76 | 4 | 26.67 | 4 | 23.53 | 0.011* |
| 1.5–3 | 51 | 75.00 | 10 | 66.67 | 6 | 35.29 | |
| > 3 | 9 | 13.24 | 1 | 6.66 | 7 | 41.18 | |
There was a statistically significant difference in the number and duration of attacks between patients with or without family history of FMF, with p < 0.001 and 0.015*, respectively (Table VI).
Discussion
Familial Mediterranean fever is an autosomal recessive diseases with specific predilection to Mediterranean areas. Kafrelsheikh is located in the north of Egypt and is one of the Mediterranean regions; despite this, to our knowledge, no previous study has been performed to assess and evaluate the pattern of FMF in Kafrelsheikh. We started the diagnosis with patients who were suggested clinically and confirmed by MEFV gene mutation analysis.
In this study, we described the distribution of 12 MEFV gene mutations. 67% were female and 33% were male, i.e. with more disease predilection in females. This is discordant with the work of Mansour et al., who found that 50.9% were males and 49.1% were females, i.e. with disease predilection in males [9]. This may relate to the total number of patients who agreed to participate.
Family history was positive for FMF in 61%, and parenteral consanguinity was found in 52%, which is slightly higher than in the study conducted by El Gezery et al. in 2010 [10].
Clinical assessment
Our data are concordant with different studies which recorded that abdominal pain is the most common presentation followed by fatigability and generalized tiredness. Karadag et al. consider generalized tiredness as a predisposing factor of the disease activity, not a presentation of the disease itself [11]. Interestingly, we found that 57% of the patients underwent abdominal surgery before the diagnosis due to the severity of the symptoms.
Attack severity and duration
Our study showed that the range of the number of attacks per year was 4–35 and the mean duration of the attacks was 36.62 h, which agrees with El Gezery et al. 2010 that the range of the number of the attacks per year was 2–32 and the mean duration of the attacks was 48 h [10]. We noticed also that the number of attacks was higher in patients with positive family history and in homozygous patients. Tchernitchko et al. indicated that M694V homozygote and heterozygote genotype is the most significant risk factor for amyloidosis [12].
Imaging study and laboratory investigation
A previous study by Amal showed that splenomegaly was present in 8.5% [9]; in our study we found that the mean level of splenic size was 12.17 ±1.35 – this was explained by affection of the reticuloendothelial system in FMF, as mentioned by Rimon et al. [13].
The E148Q mutant allele was the most commonly encountered in the studied patients from the Kafrelsheikh governorate, with a frequency of 31%, followed by M6801 (G/A), which was associated with the highest amyloid A level. These results were contradictory to Zekri et al., who described the M694I mutation as being the most frequently detected mutation in all FMF patients [14].
Response to treatment
In our study more than 50% of patients responded to a dose of 1.5–3 mg daily, and this is not in agreement with Al-Wahadneh and Dahabreh, who reported that prescribing colchicine to all of their patients in doses ranged between 0.5 mg and 2 mg daily resulted in the disappearance of the attacks completely in 68% of cases; this may be partial related to the type of gene mutation [15].
Conclusions
Patients with positive family history and those with homozygous mutation have more attacks with increased severity and higher amyloid deposition. The E148Q mutant allele was the most commonly encountered in the studied patients from the Kafrelsheikh governorate, with a frequency of 31%, followed by M6801 (G/A), which was associated with the highest amyloid A level.