








INTRODUCTION
Noonan syndrome (NS) is a disease that occurs in 1 in 1,000–2,500 live births, mostly in an inherited autosomal dominant manner. Its clinical phenotype varies according to the severity of the disease and may involve multiple organ systems throughout the patient’s lifetime [1, 2]. Noonan syndrome is a clinically and genetically heterogeneous disease that may be accompanied by growth retardation, prominent facial dysmorphic features, congenital heart defects, hypertrophic cardiomyopathy, skeletal anomalies, bleeding diathesis, ectodermal anomalies, lymphatic dysplasias, cryptorchidism, and cognitive disorders [3–5].
Noonan syndrome is a member of the RASopathy family resulting from activating mutations in genes encoding components or regulators of the RAS/mitogen-activated protein kinase (MAPK) signal transduction pathway, which is essential for cell cycle, differentiation, growth, and aging [1, 2, 6]. The spectrum of diseases in this group includes cardiofaciocutaneous syndrome, Costello syndrome, neurofibromatosis type 1, Legius syndrome, Leopard syndrome, NS-like syndrome with loose anagen hair and capillary/arteriovenous malformation syndromes [7, 8].
Some of the patients with NS may experience various clinical issues related to immunodeficiency, such as recurrent infections. This article discusses a very rarely seen case of a 15-year-old male patient with NS having common variable immunodeficiency disease (CVID).
CASE REPORT
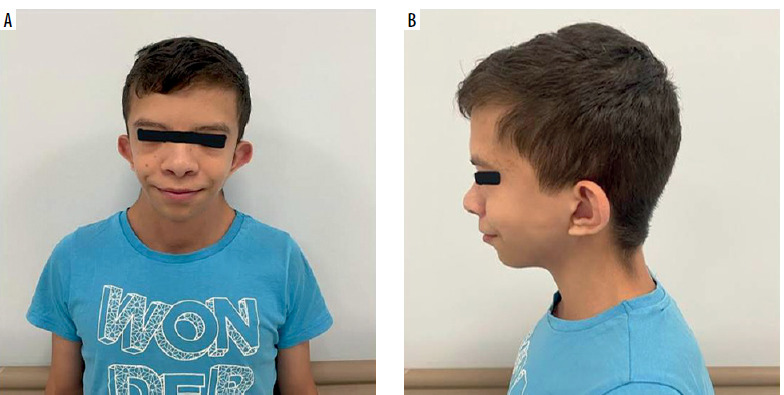
A 15-year-old male patient was followed up by the pediatric endocrinology department because of hypothyroidism and growth retardation with a diagnosis of NS (Figures 1 A, B). He presented to the pediatric allergy outpatient clinic with the complaint of urticaria plaques that had been persisting for 3 months and occurred almost every day. (Written informed consent was obtained from the patient and his parents for the patient’s presentation here.)
While the etiology of chronic urticaria was being investigated, it transpired that the patient had been frequently ill since infancy and had often mild respiratory infections. The patient had bronchitis three times in the last year and had been hospitalized three times due to high fever. Although this patient had recurrent episodes of bronchitis, they were triggered by upper and lower respiratory tract infections. He had otitis and pneumonia (one episode each). Our patient had also multiple hospitalizations, but most of them were due to frequent fevers of unknown cause and growth retardation at an early age. His parents were not related, and any member of the family had no known primary immunodeficiency disease.
The results of routine hematologic, biochemical, and hormonal tests were found to be normal and are shown in Table 1. Allergy screening was performed twice with serum specific IgE evaluation and once with a skin prick test, but nothing was detected. In the first immunological evaluation, the following was observed IgG: 573 (normal (value): 895–1108) mg/dl, IgA: 44.8 (normal: 97.8–130) mg/dl, IgM: 48 (normal: 83.7–138) mg/dl, anti-HBs: negative, anti-CMV IgG: negative, anti-rubella IgG: low positive (42 IU/ml), anti-A: 1/64, and anti-B: 1/64 (Table 2). The patient’s immunoglobulin values, repeated 1 month apart for control purposes, were IgG: 462 mg/dl and 438 mg/dl, IgA: 42 mg/dl and 44.9 mg/dl, and IgM: 27mg/dl and 42 mg/dl. No other causes of hypogammaglobulinemia were present. Flow cytometric lymphocyte subpopulation analysis showed that CD19+-cell: 19% (normal: 10–30), CD3+-cell: 63.1% (normal: 58–82), CD4+-cell: 33.6% (normal: 26–48), CD8+-cell: 23.5% (normal: 16–32), and CD16+-56+-cell: 14.4% (normal: 8–30) (Table 3). Peripheral lymphocyte subpopulation analysis showed normal percentages/absolute counts. With this clinical history, clinical symptoms/signs, and hypogammaglobulinemic laboratory results, the patient was diagnosed with CVID, and intravenous immunoglobulin (IVIG) replacement therapy was started at 0.5 gr/kg once a month. The patient received his first IVIG at a 10% concentration of 0.5 g/kg over 4 h, and no reaction was observed during infusion. The patient’s complaints about recurrent respiratory infections decreased after IVIG. The patient had significant clinical benefits from IVIG.
Table 1
Hematological, biochemical, and hormonal parameters of the patient
Table 2
Immunological (specific and non-specific antibody) evaluation of the patient
Table 3
Peripheral blood lymphocyte subset evaluation of the patient
Evaluations for chronic urticaria did not reveal any recognizable cause. Interestingly, his urticarial lesions did not recur after IVIG treatment started. Moreover, Sanger DNA sequence analysis showed p.Met504Val (c.1510 A>G) heterozygous mutation in the PTPN11 (protein tyrosine phosphatase non-receptor type 11) gene, this mutation is known to a pathogenic one for NS. This mutation was previously defined in HGMD (HGMD no: CM013423).
DISCUSSION
Immune system abnormalities or autoimmune diseases are rarely reported in patients in the RASopathy disease group, including Noonan syndrome. Common clinical features observed in NS patients are summarized in Table 3 [9]. In a cohort study of 69 patients with RASopathy and 50 controls, it was reported that IgA levels were lower in patients compared to controls. IgA deficiency was observed in 18% of the patient group [10]. Consistent with the literature, serum IgG and IgM levels were also found to be low in addition to IgA. In accordance with this laboratory result, our patient had a history of frequent illnesses, history of severe febrile infections, and frequent bronchitis.
Although NS is an autosomal dominant disorder that is characterized by dysmorphic features, short stature, and congenital heart disease, immunodeficiency has not been documented as part of the syndrome (Table 4). In the literature (abstract presentation), 1 case with NS and hypogammaglobulinemia was reported. This case reported by Hostoffer et al. with NS and hypogammaglobulinemia was a 32-month-old female patient with recurrent otitis and sinusitis. Her serum Ig G, IgA, and IgM values were low compared to the reference ranges and were probably diagnosed as transient hypogammaglobulinemia of infancy. Clinical follow-up was decided for this case; prophylactic antibiotics were started, and the patient was removed from the daycare center. With these measures, a decrease in the frequency of infection (only 1 episode of otitis media) was observed [11].
Table 4
Common clinical features of Noonan syndrome
In addition, in a retrospective study of 36 cases of primary immunodeficiency, aged 0–18 years, reported from the Bristol Royal Hospital for Children, United Kingdom, 17/36 cases with genetic syndromes were identified. One of these 17 patients had NS. This article shows that this patient with NS had antibody deficiency and therefore recurrent severe infections. This patient also seems to have an allergic disease that was not mentioned. This patient was placed on immunoglobulin replacement therapy and antibiotic prophylaxis because of antibody deficiency [12].
In NS, short stature, cryptorchidism, delayed puberty, and rarely hypothyroidism are some of the defined endocrine disorders. Short stature is a common finding and may be due to growth hormone deficiency, neurosecretory dysfunction, or growth hormone resistance [13–15]. Binding of growth hormone to its receptor activates the intracellular RAS-MAPK pathway, resulting in signal transduction including proliferation of chondrocytes in the growth plate and phosphorylation of a series of proteins that stimulate linear growth. Mutations in the RAS-MAPK pathway, as in the PTPN11 gene in our patient, cause impaired proliferation of chondrocytes in the growth plate and linear growth and short stature [16]. Although recombinant growth hormone has been approved by the FDA for NS, treatment is still controversial. Our patient who used growth hormone for 8 years had a partial response to growth hormone (mean: 6.1 cm/year). The majority of men with NS have cryptorchidism. Gonadal dysfunction has also been reported in men and it has been suggested that this is due to primary Sertoli cell dysfunction rather than cryptorchidism [17]. In our patient who was operated on for cryptorchidism and followed up, increased FSH and low inhibin B levels (35.7 pg/ml, reference range 47–383 pg/ml) during follow-up support Sertoli cell dysfunction in these patients. Although autoimmune hypothyroidism with thyroid antibodies is more common in NS, the frequency of hypothyroidism is similar in the general population [18–21].
Our case was a 15-year-old male patient with endocrinopathies. In addition to a history of frequent infections, the patient had chronic urticaria, and immunodeficiency was incidentally detected by deepening the history and laboratory evaluations when the patient was being investigated for chronic urticaria. In the follow-up of the patient, it was observed that both frequent infections and chronic urticaria regressed after IVIG treatment was started.
CONCLUSIONS
Patients with NS who experience recurrent infections may have hypogammaglobulinemia. In addition to our patient, a couple of cases of coexisting NS and primary immunodeficiency (predominantly antibody deficiency) have been reported in the literature so far. Further immunologic evaluations are needed to determine whether primary immunodeficiency is a component of the syndrome in this population.