

Introduction
Hypersensitivity pneumonitis (HP) is an increasingly recognized interstitial lung disease (ILD), usually ranked third, after idiopathic pulmonary fibrosis (IPF) and sarcoidosis or ILD during collagen tissue diseases [1-3]. The first diagnostic step of HP is to identify a symptomatic disease, which develops due to exposition to various, mostly organic, antigens [4]. The second step is to detect an interstitial lung disease with characteristic radiological features in chest computed tomography (chest CT), and the third step involves an evaluation of increased number of lymphocytes in bronchoalveolar lavage fluid (BALF) [4]. Lung biopsy should be considered in case of diagnostic difficulties [5].
According to recent suggestions of experts, two forms of HP have been distinguished: acute type, with symptoms’ duration of 6 months or less, and chronic form, with the symptomatic disease lasting for more than 6 months [4, 5]. Within the group of chronic HPs, two subgroups were described: with or without lung fibrosis [4].
HP presents with increased total cell counts and percentage of lymphocytes in BALF. Lymphocytosis of 60-80% has been described in acute phase of the disease [6, 7]. Therefore, pulmonary specialists in DELPHI study indicated BALF lymphocytosis of 40-50% as a significant factor for differentiation between HP and most other ILDs with lymphocytosis in BALF, such as sarcoidosis, non-specific interstitial pneumonitis, or cryptogenic organizing pneumonia [8].
Despite many published reports, there is no consensus regarding the changes of BALF cellular composition in patients with prolonged HP course. Most authors suggested that BALF lymphocytosis may be lower in chronic fibrosing HP than in acute non-fibrosing disease, especially in smokers [9-12]. Therefore, the value of HP diagnostic algorithm is undefined in chronic phase of the disease.
Therefore, considering the above, we decided to investigate the possible relation between BALF cells’ subpopulations and the disease duration, smoking habits, and the extent of lung fibrosis on chest CT in patients with newly recognized HP.
Material and methods
Clinical presentation, imaging studies, and laboratory testing results of 94 patients, 49 females, 45 males, mean age 52 (±12) years, with newly recognized HP, were analyzed retrospectively. The applied diagnostic criteria of HP recognition have been described previously [13, 14] and consisted of:
Identification of antigen responsible for the symptoms of disease and/or the presence of specific IgG antibodies in serum of symptomatic patients,
Characteristic pattern of interstitial lung disease on chest CT (mosaic attenuation pattern of lung parenchyma with air trapping and/or ill-defined centrilobular nodules),
Increased percentage of lymphocytes in BALF (> 30% in non-smokers and >20% in smokers),
Histopathology suggestive of HP in the specimen obtained during transbronchial or surgical lung biopsy (in cases not diagnosed with criteria 1-3).
Patients’ clinical records were reviewed retrospectively, and the time from first symptoms of the disease to the diagnosis of HP was determined in months. Acute form of the disease was recognized if the time from first symptoms to diagnosis has not been longer than 6 months, while chronic form, if the symptomatic disease lasted for more than 6 months.
Chest X-rays and chest CT examinations performed at diagnosis, with or without contrast media, on CT scanners with 16 or more rows, were subsequently reviewed by two independent radiologists with more than ten years of experience in ILD diagnostics.
Diagnostic chest CT scan required complete imaging of the lungs on inspiration and slice thickness of 1.5 mm or less. In some patients, expiration scans have also been performed.
Radiologists evaluated the presence and intensity of typical HP features in CT images, including centrilobular nodules, diffuse ground glass opacifications, air trapping, and mosaic attenuation, and features of lung fibrosis, such as reticulation, traction bronchiectasis, and honeycombing. Lung fibrosis on CT images was staged as: 0. No fibrosis (Fig. 1A), 1. Areas of reticulation with traction bronchiectasis, 2. Advanced lung fibrosis with honeycombing and bronchocentric fibrosis (Fig. 1B). Characteristics of study population is presented in Table 1.
Fig. 1
Non-fibrosing and fibrosing hypersensitivity pneumonitis. Figures present CT axial images of the lung window. A) Bilateral patchy ground-glass opacities, diffuse, poorly defined centrilobular nodules, focal regions of decreased attenuation corresponding to air trapping lobules and normal lungs, characteristic for HP mosaic attenuation, and no features of lung fibrosis (stage 0). B) Fibrosing hypersensitivity pneumonitis – patchy ground-glass opacities and diffuse reticulation with traction bronchiectasis and focal honeycombing, which are the features of advanced lung fibrosis (stage 2)
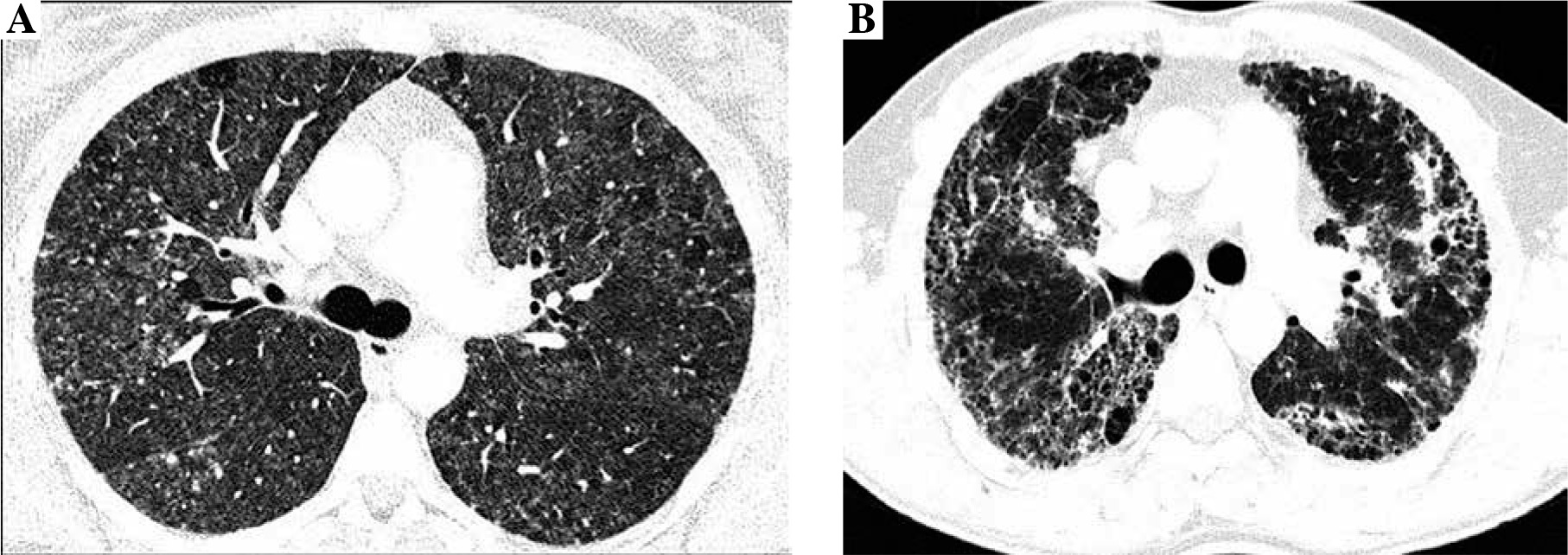
Table 1
Characteristics of the study population
BALF was performed as a part of routine testing, according to recent recommendations [15]. To collect BALF, the bronchoscope was placed in the wedge position within the bronchopulmonary segment selected during chest CT examination. Room temperature saline was instilled through a bronchoscope in 10 aliquots, up to maximum of 200 ml. The recovered BAL fluid (at least 50% of infused volume) was filtered through sterile gauze, and centrifuged (4°C, 400 × g, 15 min). Cell viability was assessed by trypan blue exclusion. Cytospined smears were stained with May-Grunwald-Giemsa differential staining and assessed by counting of a minimum 600 cells. T cells subpopulations were determined by flow cytometry using FACSCalibur TM system (Becton Dickinson, San Jose, CA, USA) and Tri TEST CD4/CD8/CD3 monoclonal antibodies (Becton Dickinson, San Jose, CA, USA) [16].
Statistical analysis
All analyses were performed with R, a software environment for statistical computing and graphics (https://www.r-project.org/). Continuous variables were presented as medians and ranges, or means and standard deviations, whereas categorical variables were described as percentages of the entire studied population. Distributions’ normality and homogeneity of variance of continuous variables in different groups were verified using Shapiro-Wilk test and F-test, respectively. If both criteria were fulfilled, Student’s t-test and Mann-Whitney U test were used for comparison of the two groups. Moreover, ANOVA test or Kruskal-Wallis test were applied to compare the three groups. Categorical variables distribution was compared with Pearson’s test, with its modifications if applicable. Correlations were analyzed using Spearman’s rank order or Pearson’s test. Proportions were compared with chi-square test; p < 0.05 was considered significant.
The project has been approved by Bioethical Committee of National Tuberculosis and Lung Diseases Research Institute (No 14/2019).
Results
Total BALF cell count was 27.4 × 106 (18.4-42.5 × 106). Median percentage of BALF lymphocytes was 48% (2.4-84.6%). In 82% of patients, BALF lymphocytosis was higher than 30%. Median of lymphocytes T CD4/CD8 index was 1.9 (0.14-48.4). The results of BALF analysis in relation to the disease duration are shown in Table 2. No significant differences were found between the two groups concerning the percentage of cells in BALF. BALF cells’ populations in relation to patients’ smoking habits are presented in Table 3.
Table 2
The results of BALF analysis (medians, ranges) in relation to the disease duration in 94 hypersensitivity pneumonitis (HP) patients
Table 3
Cells populations in BALF (medians, ranges) in relation to smoking habits of 94 hypersensitivity pneumonitis patients
No significant differences concerning BALF cells’ populations between smokers and non-smokers were observed. BALF cells’ populations in relation to the presence and the extent of fibrotic changes in chest CT are submitted in Table 4.
Table 4
Cells populations in BALF (medians, ranges) in relation to the presence and extent of lung fibrosis in 94 hypersensitivity pneumonitis patients
The total cell count and relative number of lymphocytes in BALF were significantly lower in patients with lung fibrosis stage 1 and stage 2 comparing to those without lung fibrosis (stage 0) (Table 4, Fig. 2A). Lymphocytosis exceeding 30% was noticed in all patients with no radiologic signs of lung fibrosis, 77% of patients with stage 1 lung fibrosis, and 68% of patients with stage 2 lung fibrosis.
Fig. 2
BALF results in relation to the presence and extent of lung fibrosis in patients with hypersensitivity pneumonitis. A) The percentage of BALF lymphocytes in the groups with no lung fibrosis (stage 0), with lung fibrosis presenting as diffuse reticulation and traction bronchiectasis (stage 1), and with lung fibrosis presenting as diffuse reticulation, traction bronchiectasis, and focal honeycombing (stage 2). B) The proportions of T lymphocytes CD4/CD8 in the groups with no lung fibrosis (stage 0), with lung fibrosis presenting as diffuse reticulation and traction bronchiectasis (stage 1), and with lung fibrosis presenting as diffuse reticulation, traction bronchiectasis, and focal honeycombing (stage 2)

CD4/CD8 index was the highest in case of patients with no signs of lung fibrosis, while the lowest index was observed in patients with advanced lung fibrosis; the differences between the three examined groups were significant (Table 4, Fig. 2B).
The correlations between BALF lymphocytosis and the disease duration, the degree of lung fibrosis as well as plethysmographic lung volumes and TLCO are presented in Table 5.
Table 5
Correlations between BALF lymphocytosis (%) and disease duration, lung volumes on plethysmography, TLCO, and stage of fibrosis on chest CT
BALF lymphocyte count was significantly correlated with the stage of lung fibrosis on chest CT as well as the percentage of predicted total lung capacity (TLC) and maximal vital capacity (VCmax), but not with lung transfer capacity for carbon monoxide (TLCO). An inverse correlation between disease duration and BALF lymphocyte count was observed.
Discussion
BALF is an easily performed and well tolerated procedure, with proven diagnostic role in interstitial lung diseases, such as acute and chronic eosinophilic pneumonia, pulmonary alveolar proteinosis, pulmonary Langerhans histiocytosis, and diffuse alveolar hemorrhage [6, 15].
BALF lymphocytosis has been listed as one of important diagnostic criteria of HP, but the results of clinical studies indicate its greatest utility in the acute phase of the disease, and less certain – in chronic HP [17]. Large study conducted in Japan in chronic bird-related and home-related HP, documented lack of increased lymphocyte counts in many cases [18]. Therefore, one of the main issues we addressed in the present study was the relation between the disease duration and BALF cellular composition in patients with newly recognized HP.
Our study group included 19% of patients with acute HP and 81% of cases with chronic form of HP. We found significant inverse correlation between BALF lymphocytosis and disease duration. However, the median percentages of BALF lymphocytes in acute HP (≤ 6 months of symptoms duration) was comparable with chronic form of the disease (> 6 months of symptoms duration). Such a discrepancy may be explained by relatively high BALF lymphocyte counts in the two forms of HP (values exceeding 30% were found in 88% of those with acute HP and 68% of those with chronic HP). In our opinion, the demonstration of increased percentages of lymphocytes in many patients with prolonged course of HP was the proof of high diagnostic value of BALF examination, even in patients with chronic form of the disease.
Another factor, which potentially may influence BALF composition is cigarettes smoking. According to literature data, an increased total BALF cells’ count and alveolar macrophages have been observed in healthy smokers [6, 19]. In smokers with HP, BALF lymphocyte counts may be lower comparing to non-smokers [20]. Therefore, many authors suggest that BALF lymphocytosis exceeding 20% should be used as a diagnostic threshold of lymphocytic alveolitis in smoking HP patients, as it corresponds to 95% of values observed in a healthy smoking population [7, 9]. Our study group included 41% of smokers, but we did not observe any differences concerning BALF cells populations between smokers and non-smokers.
The most important finding of our study group was a significant inverse correlation between the degree of lung fibrosis on chest CT and BALF lymphocytosis (r = –0.59, p = 0.0001). The more advanced lung fibrosis, the lower median BALF lymphocytosis observed. Therefore, it was not surprising that BALF lymphocytosis was significantly correlated with plethysmographic lung volumes (TLC% pred and VCmax% pred).
BALF lymphocyte counts exceeding 30% were found in 100% of patients with no fibrotic lung lesions on chest CT, 77% of those with stage 1 fibrosis, and 68% of those with stage 2 fibrosis. If the higher cut-off were used (e.g., 50% as proposed by DELPHI study), the proportion of patients with elevated BALF lymphocytosis would be 72%, 23%, and 15%, respectively. Therefore, we suggest using a cut-off of 30% for BALF lymphocytosis when diagnosing HP in patients with fibrotic lung disease. Applying higher cut-off values could markedly decrease the sensitivity of the test. Another possibility suggested by Adams et al. was to increase the diagnostic value of BALF in HP and combining it with trans-bronchial lung biopsy, especially in cases with fibrotic HP, where a lower lymphocyte counts are expected [21].
When the diagnosis of usual interstitial pneumonia (UIP) is considered as alternative to fibrotic HP, the used threshold of BALF lymphocytosis may be even lower than 30%, due to normal BALF lymphocytic count in most UIP patients [22]. Tsilas et al. found BALF lymphocytosis exceeding 20% in 35 out of 95 patients with non-classified fibrosing lung diseases diagnosed as indeterminate UIP, and one third of them have been subsequently classified as chronic fibrosing HP [23].
In our study, median CD4/CD8 index was 1.9. According to some literature, lymphocytosis in BALF with CD4/CD8 index lower than 1 should be suggestive for HP [24, 25]. However, other authors questioned the diagnostic importance of CD4/CD8 in HP [26, 27]. Caillaud et al. found CD4/CD8 < 1 in 30% of HP patients only [27].
Vasakova et al. speculated that proportion of CD4/CD8 in BALF of HP patients may depend on type of antigen and intensity of exposition as well as the time span between the exposition and BALF collection [20]. In recently described Portuguese cohort of HP patients, CD4/CD8 ratio was significantly lower in patients with acute HP comparing to the remaining ones [28]. It is possible that an increase of CD8 count, which was observed after an acute exposition to antigen has a protective role against lung fibrosis [20, 26]. The relative shift in balance to CD4, due to decrease of CD8 count in chronic HP, may favor pro-fibrotic mechanisms [26]. We did not find the differences of CD4/CD8 ratio between acute and chronic form of HP, but surprisingly, we found significantly lower CD4/CD8 ratio in patients with lung fibrosis comparing to those without fibrotic lung disease. The clinical value of this observation is uncertain, due to small sample size, retrospective character of the study, and discrepancies in CD4/CD8 ratio observed in the study group. Therefore, a prospective evaluation performed in larger group of HP patients is required to assess the clinical role of CD4/CD8 ratio as a possible marker of fibrotic lung disease in HP patients.
Conclusions
In our study group of newly recognized HP patients, BALF lymphocytes’ count correlated with the extent of lung fibrosis and plethysmographic lung volumes, but it was not associated with TLCO and smoking. The inverse correlation was found between the duration of symptomatic disease and BALF lymphocyte count. The role of CD4/CD8 index measurement as a marker of fibrotic lung disease requires further investigation.