










Introduction
Malaria remains one of the most devastating infectious diseases affecting millions of people every year across the globe (Kalita et al., 2020). Plasmodium falciparum, the deadliest malarial parasite, is responsible for most of the mortality associated with malaria infection (Kalita et al., 2019; Rudrapal et al., 2018). Artemisinin-based Combination Therapies (ACTs) are currently the frontline treatments against P. falciparum malaria (Patowary et al., 2019; Medhi et al., 2018; Gogoi et al., 2016; Rudrapal et al., 2016a). Despite the significant progress made in drug discovery and development over the past few decades, the control and prevention of malaria are still a challenging task. Because of the widespread emergence of drug-resistant strains of P. falciparum, the clinical utility of existing drug therapies including ACTs in the treatment of malaria has been increasingly limited (Roy et al., 2013; Rudrapal et al., 2013; Cumming et al., 1998; Neil et al., 2005). It has become a serious health concern which, therefore, necessitates the development of novel drug molecules and/or alternative therapies to combat, particularly resistant P. falciparum malaria.
Artemisinin (ART) is a naturally occurring antimalarial lead molecule effective against P. falciparum malaria. The 1,2,4-trioxane moiety is believed to be the key pharmacophore of ART and its semi-synthetic derivatives (Posner, 1997; Chakroborty et al., 2012; Opensica et al., 2009; Rudrapal et al., 2019). ART exhibits antimalarial action by forming highly alkylating free radicals and reactive oxygen species after interaction with iron (Rudrapal et al., 2017; Chetia et al., 2016). The 1,2,4-trioxane moiety containing the peroxide functional group is cleaved during the erythrocytic stage of parasite growth, leading to the release of reactive oxygen radicals that kill the parasite by peroxidation of parasitic proteins or cell membrane (O’Neill et al., 2005; Wang et al., 2009; Robert et al., 2020; Biamonte et al., 2013; Mugumbate et al., 2013). The objective of the present study was to develop 1,2,4-trioxane derivatives as novel antimalarial agents that would be effective against resistant P. falciparum. In our study, 15 new trioxane derivatives were designed as possible antimalarial agents by molecular modification of the 1,2,4-trioxane scaffold. Molecular modeling studies of trioxane derivatives were performed based on the CADD approach using Biovia Discovery Studio (DS) 2018 software. The protein-ligand docking study was conducted against P. falciparum cysteine protease FP-2 (PDB ID: 3BPF) protein using the simulation-based docking protocol LibDock by the flexible docking method. The assessment of drug-likeness, ADMET properties, and toxicity was also performed. Further, molecular dynamics (MD) simulation studies were performed along with the calculation of MM-PBSA-based free binding energies of docked complexes to confirm the antimalarial potential of the best scored compounds against the P. falciparum FP-2 enzyme. Additionally, the selectivity of trioxane hits identified as potential inhibitors of P. falciparum cysteine protease FP-2 was determined on human cysteine proteases such as cathepsins (Cat K and Cat L), which are the host homologous proteins.
Materials and methods
Design of new 1,2,4-trioxane derivatives and molecular modeling studies
In this study, a 1,2,4-trioxane scaffold was used as the basic structural framework for designing new trioxane derivatives by the molecular modification approach. Fifteen trioxane derivatives were designed by the substitution of the aryl moiety (substituted biphenyl-3-carbaldehyde) at C3 and 4-fluorobenzoyl moiety at C6 as depicted in Fig. 1. The trioxane ring with a bulk aryl substituent containing an aldehydic group attached at the 3rd position could serve as an important factor for their antimalarial efficacy.
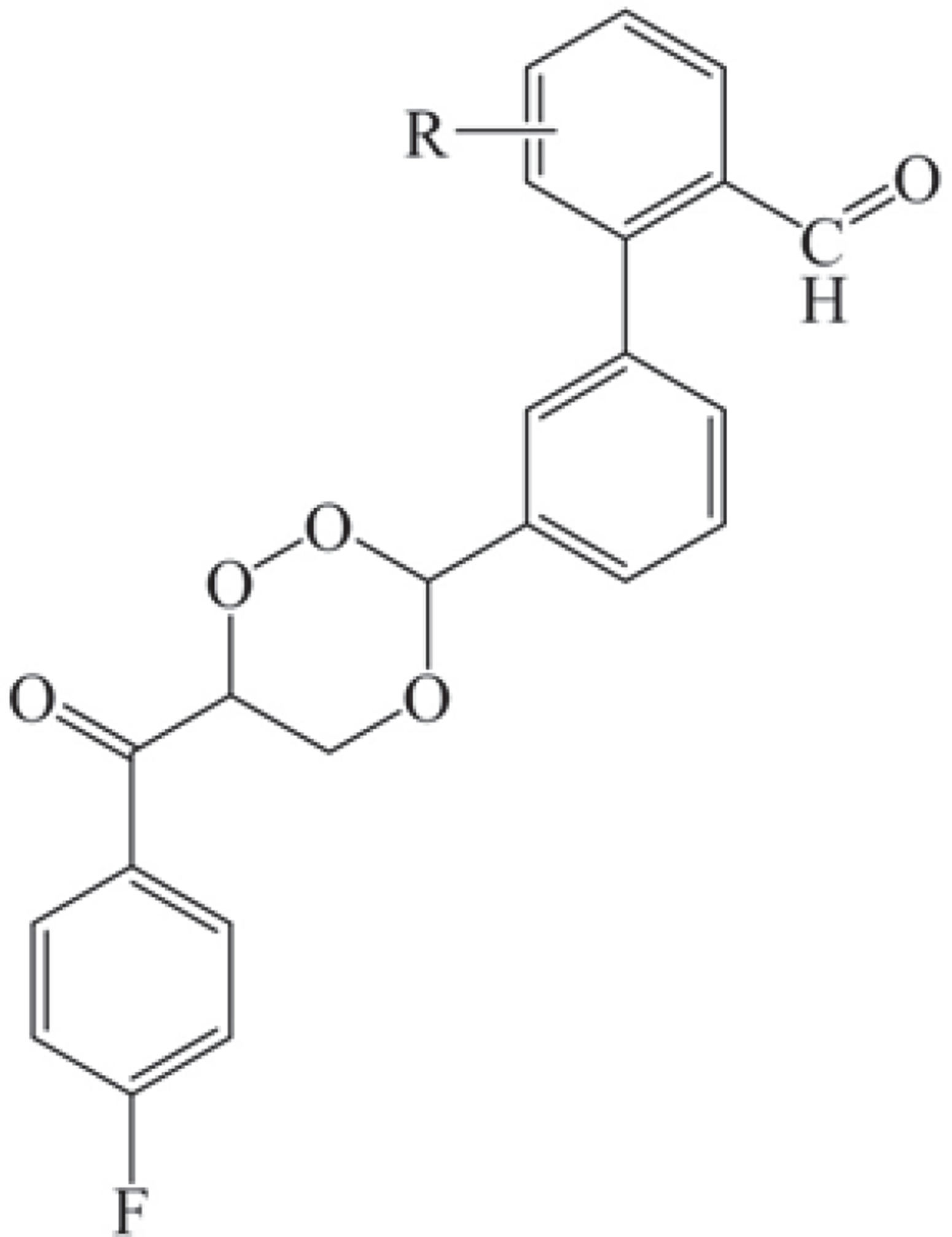
Molecular modeling studies were performed on Biovia Discovery Studio (DS) 2020 software (Dassault Systèmes BIOVIA, San Diego, USA) by using Dell Precision workstation T3400 running on Intel Core 2 Duo Processor, 4 GB RAM, 250 GB hard disk, and NVidia Quodro FX 4500 graphics card.
Molecular docking study
Prior to docking, ligands and proteins were prepared according to the standard protocol of DS software. The three-dimensional structures of compounds were generated, and their energy minimization was performed using the CHARMM (Chemistry at Harvard Macromolecular Mechanics)-based smart minimizer. It performs 2000 steps of Steepest Descent followed by the Conjugate Gradient algorithm with an RMSD gradient of 0.01 kcal/mol (Mugumbate et al., 2013). The X-ray crystal structure of FP-2 (PDB ID: 3BPF) protein was retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/). Chain A of 3BPF determined at the resolution of 3.2 Å was used in the study (Ghorab et al., 2020; Abdel-Hamid et al., 2014). The protein molecules were prepared by necessary cleaning, removal of water molecules, and energy minimization following the standard protocol. The energy minimization was performed using the CHARMM-based smart minimizer method at the maximum steps of 200 with RMSD gradient of 0.1 kcal/mol (Jain et al., 2008).
A receptor grid was generated around the binding cavity (active sites) of the energy-minimized protein molecule by specifying the key amino acid residues. The co-crystal ligand (active inhibitor) of the respective protein molecule was selected to define the amino acid residues for predicting the binding sites. For the generated receptor grid boxes, the binding site sphere was set with the radius of 2.90 Å and x, y, z dimensions of 10.858743, 12.558267, 85.699954 for 3BPF (Ponnan et al., 2013; Rudrapal et al., 2017).
The docking was performed using a simulation-based docking protocol, i.e., LibDock of DS 2020. LibDock is a high-throughput docking algorithm used to dock compounds into the active site(s) of a receptor molecule. All docking and consequent scoring parameters used were kept at their default settings (Kashyap et al., 2016; Sharma et al., 2016). LibDock docked compounds were estimated. All docked poses were scored and ranked. The binding affinities of docked compounds were predicted by analyzing interactions of receptor-ligand complexes. The binding modes of the best docked pose were analyzed using the 3D receptor-ligand complex. Different nonbonding interactions (hydrogen bonding, hydrophobic, etc.) were also analyzed using the 2D diagram of receptor-ligand complexes. The docking results of test compounds were compared with those of the co-crystal ligands. The docking energies of the co-crystal ligands were used as reference ligands to evaluate the binding affinity of the test compounds.
Drug-likeness studies
The molecular properties and drug-likeness parameters of the compounds CC1–CC15 were calculated based on theoretical approaches using DS 2020. Molecular properties such as log of octanol/water partition coefficient (LogP), molecular weight (MW), number of hydrogen bond acceptors (nHBAs), number of hydrogen bond donors (nHBDs), and molecular polar surface area (PSA) incorporated in Lipinski’s rule of five (Rudrapal et al., 2019) and other physicochemical parameters such as number of aromatic rings (nARs), number of rings (nRs), and number of rotable bonds (nRotBs) were also calculated. The relatively higher lipophilicity of the target compounds could account for the good antimicrobial activities due to the increased intracellular concentration. The Molinspiration online software (http:/www.molinspiration.com/, 2016) was used to determine non-violation of drug-likeness and drug-likeness score.
ADMET prediction
The ADME-Toxicity (ADMET) parameters were calculated using the ADMET descriptor protocol of DS 2020 software. Six mathematical models (aqueous solubility, blood-brain barrier penetration, cytochrome P450 (CYP) 2D6 inhibition, hepatotoxicity, intestinal absorption, and plasma protein binding) were used for quantitative prediction of properties related to ADMET characteristics or pharmacokinetics (PKs) of drug molecules (Rudrapal et al., 2019).
Toxicity assessment
In silico toxicities were assessed using the TOPKAT module of DS 2020 software (Rudrapal et al., 2019). The following toxicity parameters were calculated to assess carcinogenicity and mutagenicity: mouse female NTP, mouse male NTP, rat female NTP, rat male NTP, rat oral LD50, mouse female FDA, mouse male FDA, rat female FDA, rat male FDA, Ames prediction, and DTP prediction .
MD simulation
The compounds CC3 and CC7 that showed the best binding affinities against the target protein P. falciparum FP-2 were further considered for the MD simulation study by using DS 2020. The protein-ligand complexes generated from the binding affinity prediction analysis were subjected to the MD simulation study along with the original crystal structure of the target proteins complexed with the co-crystal inhibitors. The protein-ligand complexes were initially cleaned and prepared using the macromolecule tool of DS 2020 (Lin et al., 2011). The complexes were solvated using the explicit periodical boundary condition in a cubic box of water with a distance of 5 Å from the boundary. The system was neutralized by adding 0.15 M NaCl during the solvation process. The solvated systems were energy minimized (5000 steps steepest descent and 5000 steps conjugate gradient with energy RMSD gradient 0.01 kcal/mol), heated (20 ps), and equilibrated (500 ps) using the “Standard Dynamic Cascade” protocol of DS 2020. Subsequently, 30 ns production was run in NVT ensemble at 300K for the whole protein-ligand complexes, where snapshots were saved at every 2 ps. For the electrostatic calculations, the Particle Mesh Ewald (PME) method and to constrain bonds containing hydrogen the SHAKE algorithm were used with the time step of 2 fs. After completing the simulation, RMSD (root mean square deviation), RMSF (root mean square fluctuation), and ROG (radius of gyration) were computed by taking the starting structure as a reference to evaluate the conformational changes of the protein-ligand complexes. Throughout the simulation period, the distances of different hydrogen bonds formed were also monitored and analyzed. Finally, different nonbond interactions were also analyzed from the average interaction of the protein-ligand complexes and compared with the interactions obtained from the starting structures (Noha et al., 2017; Lai et al., 2011).
Molecular Mechanics-Poisson Boltzmann Surface Area (MM-PBSA)-based binding free energy calculation
After MD simulation, the binding free energies for each protein-ligand complex were calculated using “Binding Free Energy-Single Trajectory” protocol of DS 2020 with the application of the MM-PBSA method. In the analysis, the binding free energies of all the generated conformations were calculated, and finally, the average binding free energy (ΔG) was determined for each protein-ligand complex.
Docking for selectivity study
To determine the selectivity of best hits for 1,2,4-trioxanes identified as potential inhibitors of P. falciparum cysteine protease FP-2 against human cysteine proteases, protein-ligand docking was performed with the identified trioxanes CC3 and CC7 against human homologous cysteine proteases such as cathepsins (Cat K and Cat L). The X-ray crystal structures of Cat K and Cat L (PDB IDs: 3OVZ and 3OF9, respectively) proteins were retrieved from the RCSB Protein Data Bank. The proteins were obtained at the resolution of 2.02 Å and 1.76 Å, respectively. A previously reported method was used in the docking study. The simulation-based docking protocol LibDock was used to perform the docking study using the DS 2020 software. All parameters for grid generation and docking analysis were kept at the default settings of the software.
Results
Docking study
The protein model (3BPF/E64) was validated and used for the docking study. Prior to docking, the receptor grid models were generated and optimized in terms of binding site spheres to achieve predictive interactions between receptor molecules and the test compounds (Fig. 2). The reference co-crystal ligand (E64) was successfully re-docked to the predicted active sites of protein molecule (3BPF) with an acceptable RMSD value of 3.529 Å. The validation study was performed to reproduce the results of ligand binding modes observed experimentally in protein-ligand complexes (Kalita et al., 2019). Results of the validation study confirmed experimental binding modes/conformations of the co-crystal inhibitor E64 in the binding pocket of the respective protein molecule 3BPF with predictable protein-ligand interactions (Fig. 3).
Fig. 2
Receptor grid model of P. falciparum FP-2 (3BPF); the figure indicates the generation of binding site/active site sphere/space in/around the FP-2 protein molecule for protein-ligand docking
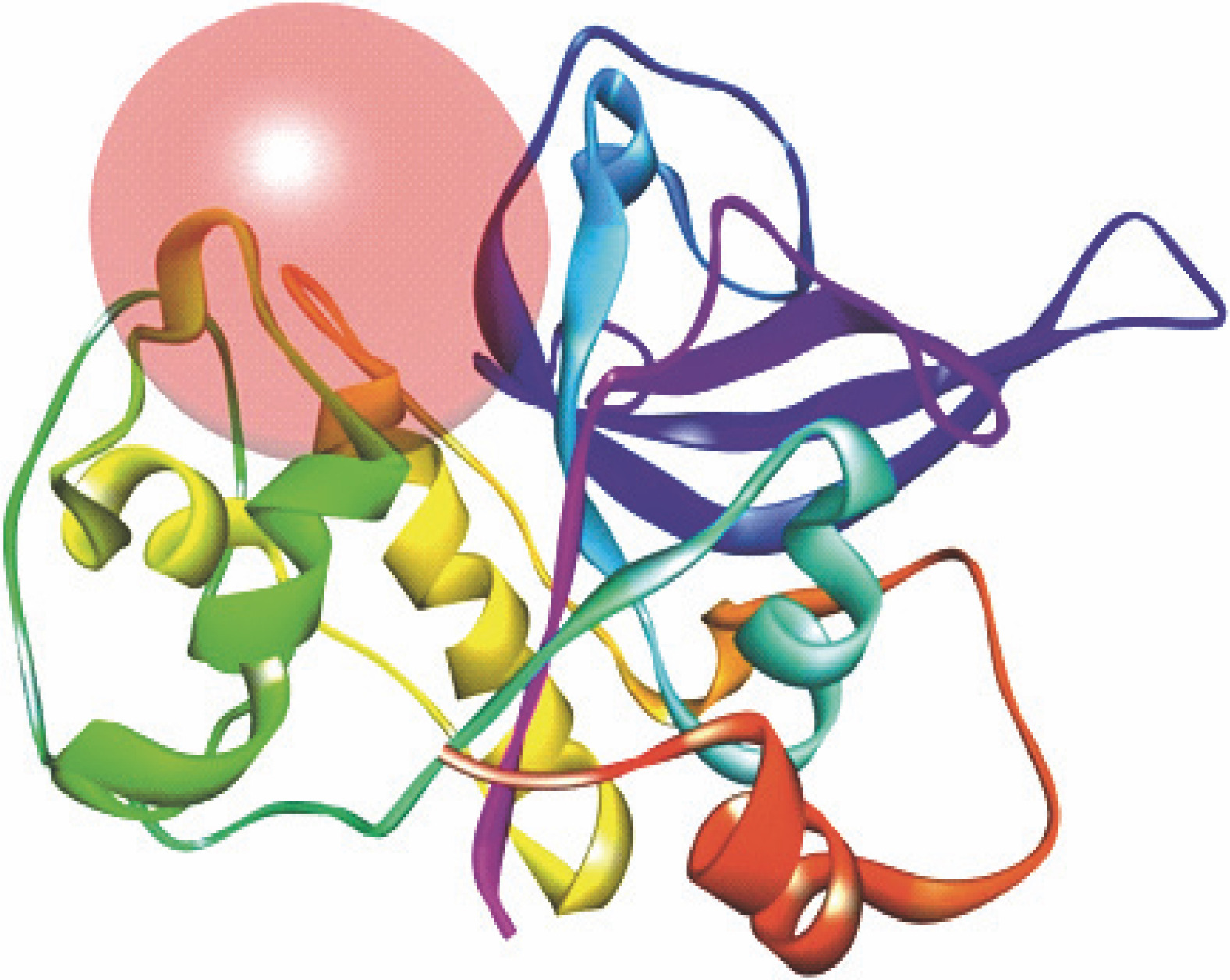
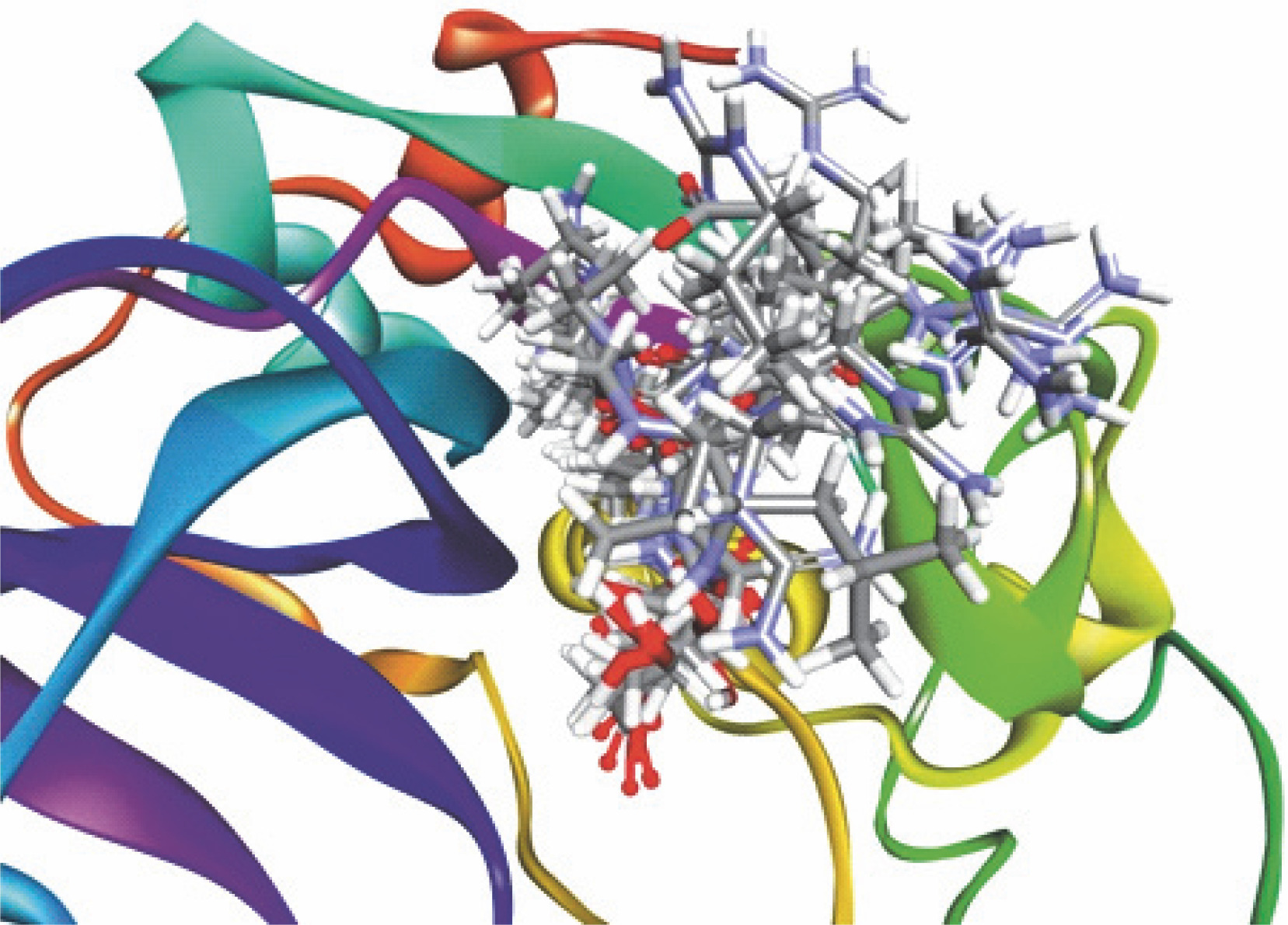
Fig. 3
Re-docked conformer of P. falciparum FP-2 (3BPF); the figure indicates re-docking of the co-crystal ligand E64 into the predicted binding pocket of FP-2; predictable binding modes were observed experimentally in the active sites of FP-2 for all the test 1,2,4-trioxane derivatives; all the test compounds and redocked E64 are superimposed into the predicted binding sites of the original co-crystal structure (E64) in the FP-2 protein
All the 15 compounds (Fig. 1) showed almost similar binding affinities with some degree of variations. Against P. falciparum FP-2 (3BPF), compounds CC3, CC5, CC7, and CC8 exhibited comparatively better inhibitory activity than the remaining analogues. Results of molecular docking are summarized in Table 2.
Table 1
Designed 1,2,4-trioxane derivatives CC1–CC15
| Compounds code | R1 |
|---|---|
| CC1 | H |
| CC2 | 4-F |
| CC3 | 4-Br |
| CC4 | 4-CH3 |
| CC5 | 4-OH |
| CC6 | 4-Cl |
| CC7 | 4-NO2 |
| CC8 | 3-OH |
| CC9 | 3-CH3 |
| CC10 | 3-Cl |
| CC11 | 3-Br |
| CC12 | 3-NO2 |
| CC13 | 2-Cl |
| CC14 | 2-CH3 |
| CC15 | 2-Br |
[i] CC – compound code (refer Fig. 1)
Table 2
LibDock scores of the designed 1,2,4-trioxane derivatives CC1–CC15 against P. falciparum FP-2 (3BPF)
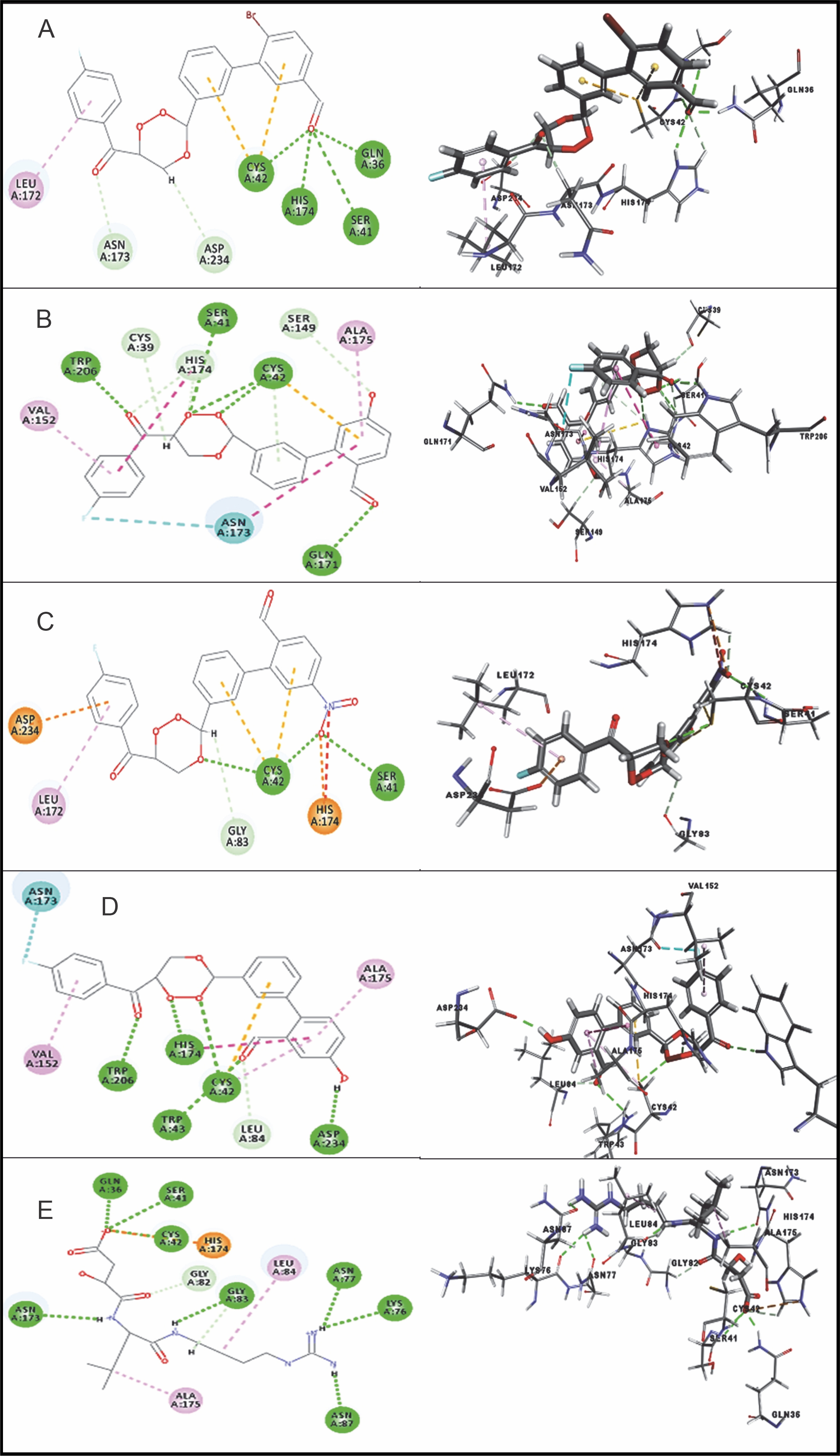
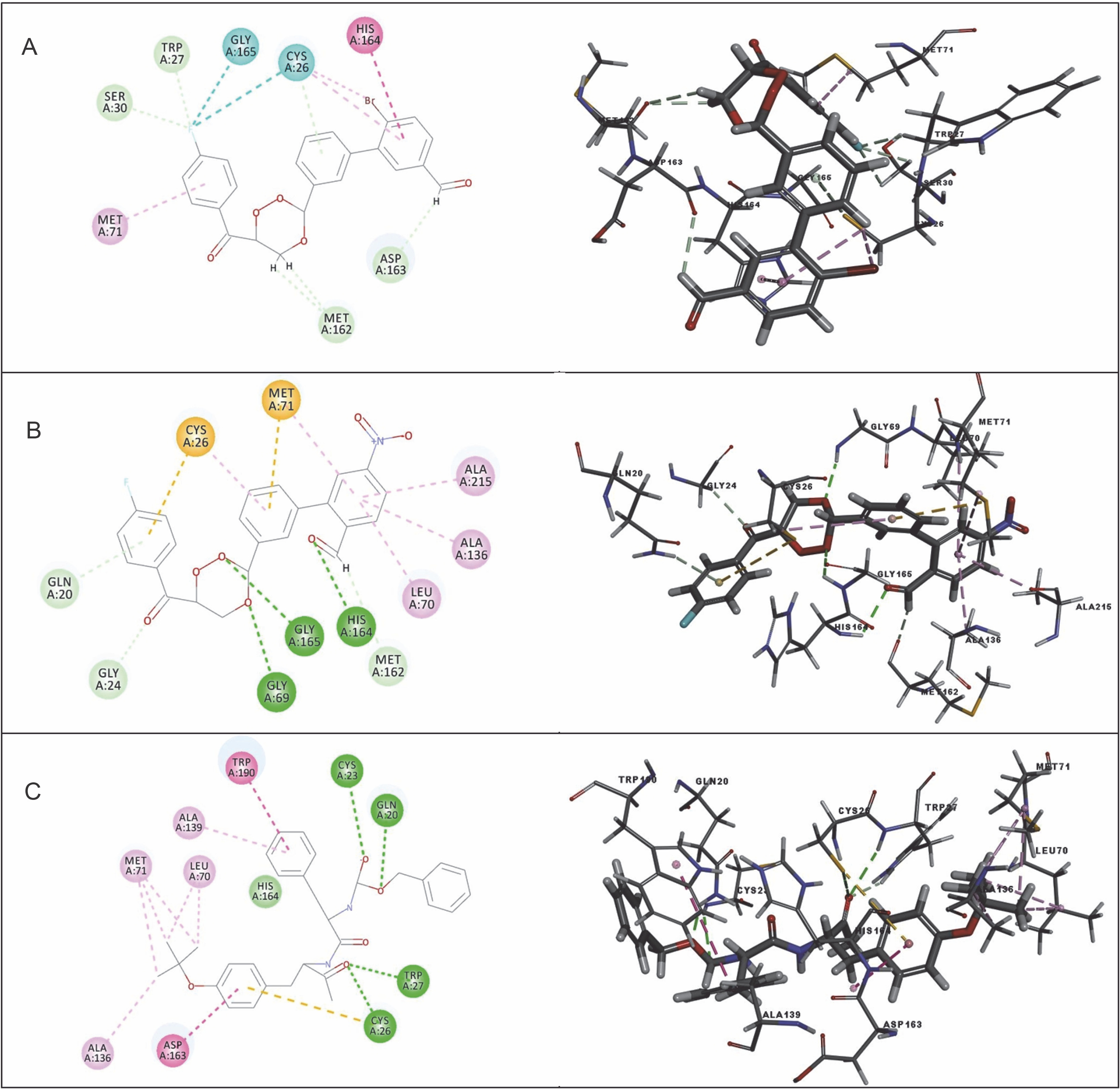
Among the 15 compounds, the compounds CC3 and CC7 showed the highest binding affinity against FP-2 with the LibDock score of 117.166 and 117.200, respectively. The Libdock score of the co-crystal inhibitor was 114.474. These two compounds were found to exhibit predictable binding affinity against the P. falciparum cysteine protease. The 3D poses of protein-ligand complexes revealed predictive binding affinity of compounds relative to the molecular orientation of receptor molecule(s). Further analysis of the 2D interaction diagram indicated that polar hydrogen bonding interactions were primarily involved between the receptor and ligand molecule, along with some secondary interactions such as hydrophobic interactions. Well-defined molecular interactions between the binding site residues of receptor molecule(s) and complementary moieties/atoms of ligands were observed. The higher the number of hydrogen bonds, the higher was the binding affinity. Other nonbonded interactions such as hydrophobic bonding were also observed, but to a lesser extent. Four potent compounds, namely CC3, CC5, CC7, and CC8, interacted with different active site residues such as Gln 36, Ser 41, Cys 42, Trp 43, Gln 171, His 174, Trp 206, Asp 234, and Trp 43 predominantly by hydrogen bond formation. These compounds possess a substituted aromatic aldehyde with –Br group at position 4, –OH group at position 3, –NO2 at position 4, and –OH at position 4. The compounds CC3 and CC7 exhibited well-defined protein-ligand interactions with very good binding modes with the formation of 7 and 5 hydrogen bonds with the FP-2 protein molecule, respectively. The co-crystal ligand formed 11 hydrogen bonds. The details of 3D binding modes and 2D interaction diagrams of four active compounds and the co-crystal inhibitor E64 are shown in Fig. 4A–E.
Fig. 4
Binding modes and docking interactions diagrams of compounds CC3 (A), CC5 (B), CC7 (C), CC8 (D), and the co-crystal inhibitor E64 (E) (left: 2D interaction, right: 3D interaction) against P. falciparum FP2 (3BPF); dotted green lines indicate conventional H-bonding interactions, and dotted yellow and purple lines indicate different types of hydrophobic interactions
Drug-likeness
The results of the calculated molecular properties and the predicted Lipinski’s parameters are shown in Table 3. The results revealed that all the compounds showed good drug-like properties based on Lipinski’s rule of five with additional parameters of drug-likeness. In our study, all the compounds exhibited satisfactory molecular properties and Lipinski’s parameters. The parameters LogP, MW, and molecular PSA indicate good membrane permeability, intestinal absorption, and oral bioavailability, respectively, whereas the other parameters nHBAs, nHBDs, nR, and Rotb bonds facilitate to produce well-defined drug-receptor interactions for optimal drug action (Rudrapal et al., 2017). None of the compounds violated the Lipinski’s rule of 5. Drug-likeness scores were recorded as 0.55 for all the designed compounds. The drug-likeness scores were within the acceptable range (0–1).
Table 3
Calculated molecular properties and drug-likeness parameters of the designed 1,2,4-trioxane derivatives CC1–CC15
ADMET
The predicted ADMET data are presented in Table 4. All the compounds (CC1–CC15) showed low to very low aqueous solubility and exhibited very poor blood–brain barrier (BBB) penetration, thereby indicating less probability of inducing CNS toxicity. The compounds were found to be both inhibitors (CC1–CC3, CC5, CC6, CC8, CC10, CC11, CC12, CC13, and CC15) and non-inhibitors (CC4, CC7, CC9, CC12, and CC14) of cytochrome P450 2D6 (CYP 2D6). The CYP 2D6 enzyme is one of the important metabolic enzymes involved in drug metabolism (Rudrapal et al., 2016b; Wang et al., 2009). The predictive hepatotoxicity was considerably observed for all the compounds. The intestinal absorption levels were observed in the range from good to very good. Plasma protein binding (PPB) data revealed that all the compounds were highly protein bound. The ADMET plot is shown in Fig. 5.
Table 4
Predicted ADMET properties of the designed 1,2,4-trioxane derivatives CC1–CC15
[i] AS (aqueous solubility) level: 2 – low, 1 – very low; BBB (blood–brain barrier) penetration: 4 – very low, 1 – high; CYP (cytochrome) P450 2D6 inhibition: true – inhibitor, false – non-inhibitor; HEPTOX (hepatotoxicity): true – toxic; IA (intestinal absorption) level: 0 – good, 1 – moderate, 2 – high; PPB (plasma protein binding): true – highly bound, false – poorly bound
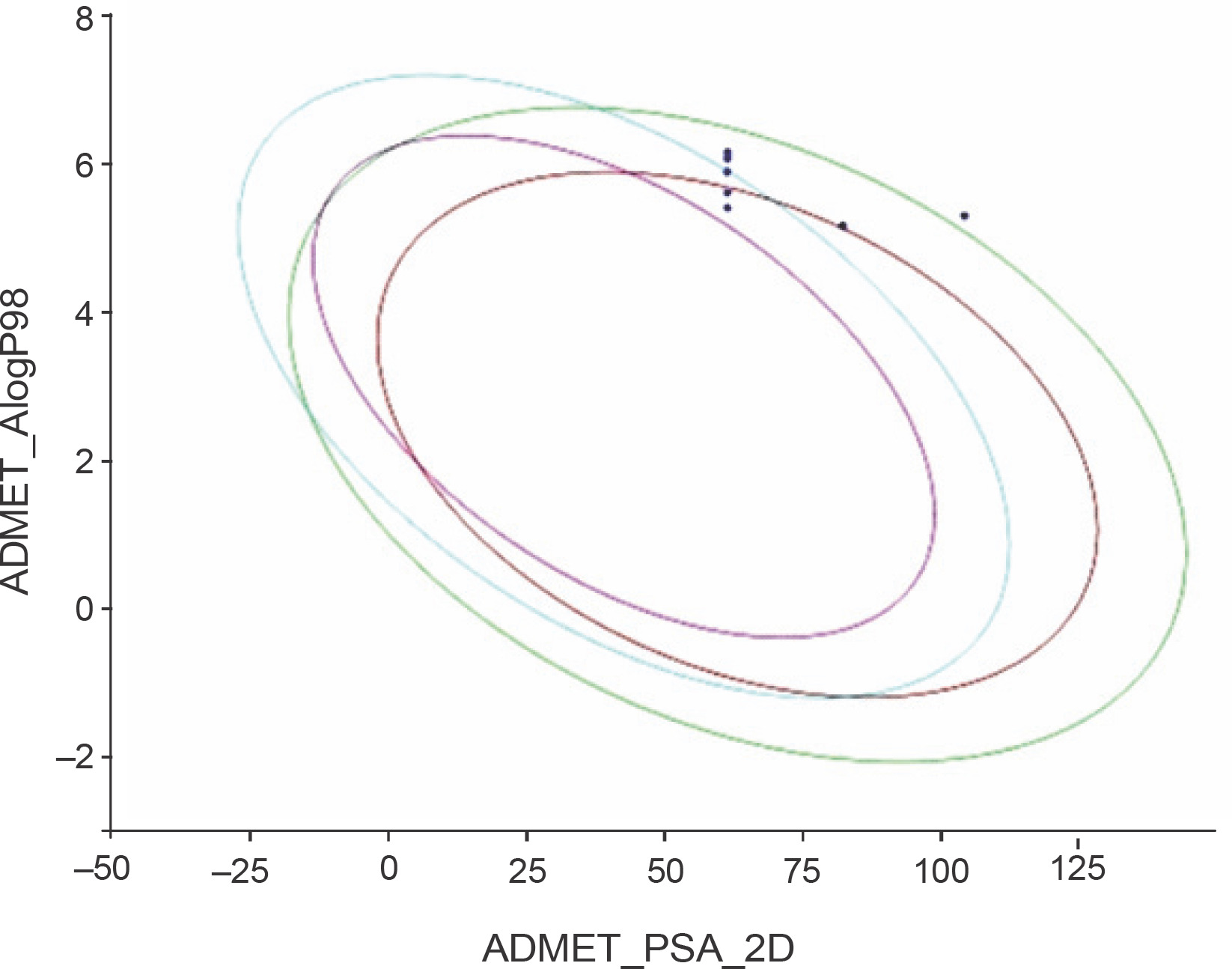
Fig. 5
ADMET plot of the designed 1,2,4-trioxane derivatives CC1 to CC15; the figure indicates that all the test compounds have acceptable ADMET characteristics; different colored lines represent different ADMET properties, while blue dots represent the test compounds with ADMET properties within the specified range
Toxicity
The toxicity data shown in Table 5A and Table 5B revealed that all the designed compounds were noncarcinogenic and nonmutagenic, except for a few ones (CC9, CC11, CC12, CC14, and CC15). Compounds CC3 and CC7 were found to be noncarcinogenic and nonmutagenic. They were found to be safe and nontoxic in various in silico models of nonruminant animals.
Table 5A
TOPKAT toxicity data of the designed 1,2,4-trioxane derivatives CC1–CC15
Table 5B
TOPKAT toxicity data of the designed 1,2,4-trioxane derivatives CC1–CC15
MD simulation
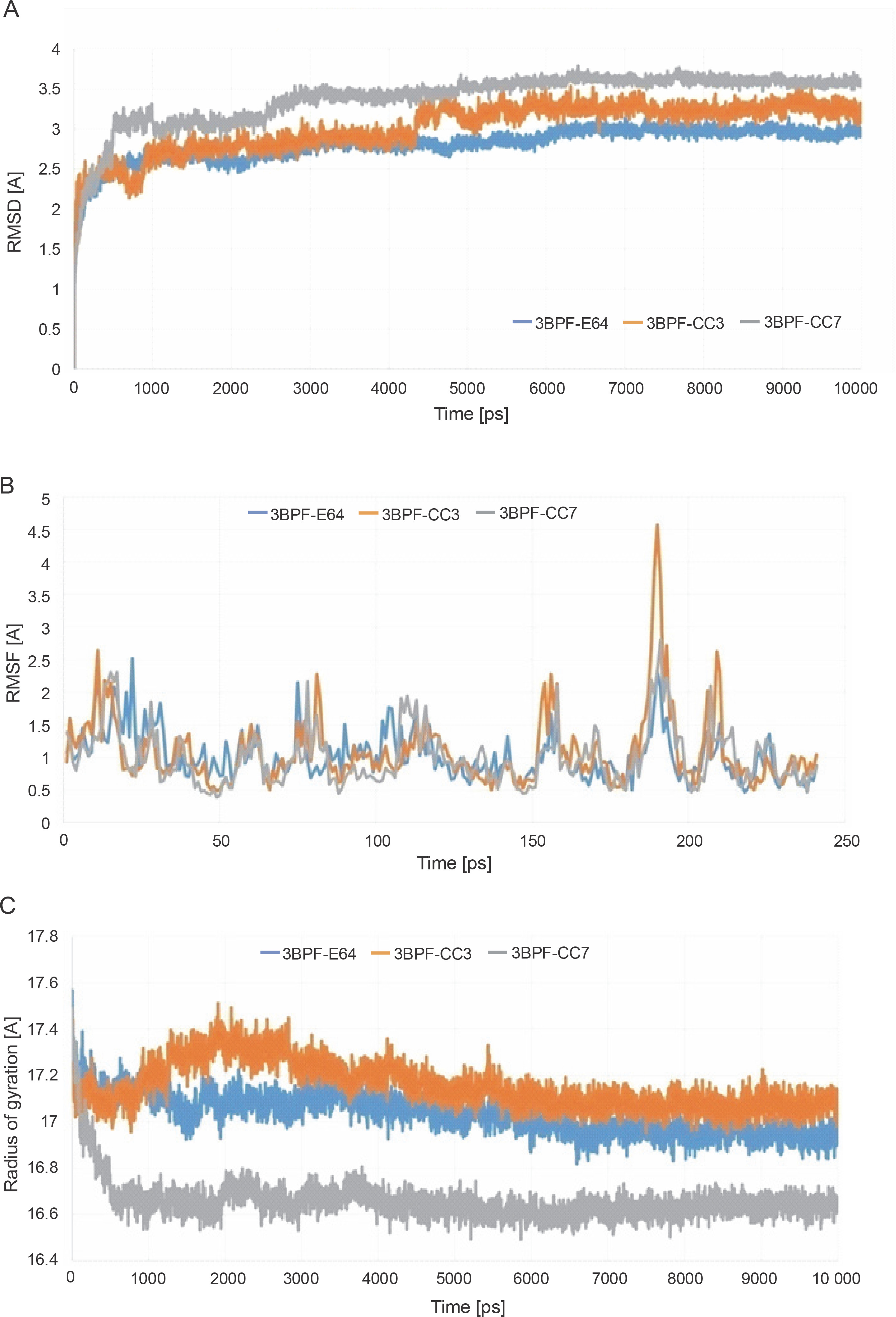
After completing the MD simulation of 10 ns, it was observed that the protein-ligand complexes formed by both the compounds CC3 and CC7 with the co-crystal ligand E64 reached the plateau states within 1 ns (Fig. 10). Within the 10 ns of simulation, the 3BPF-E64 complex deviated less than the complexes formed by the other two test compounds (CC3 and CC7), but the differences between their deviations were very small and nonsignificant (Fig. 6). The ROG diagram indicated that the 3BPF-E64 and 3BPF-CC3 complexes were formed with almost equal compactness, but CC7 formed a more compact and tighter complex with FP-2 (3BPF) than the other two complexes (Fig. 6).
Fig. 6
Stability indicating parameters obtained from the MD simulation analysis A) RMSD values, B) RMSF values, and C) ROG values; the blue line indicates the 3BPF-E64 complex, the orange line indicates the 3BPF-CC3 complex, and the gray line indicates the 3BPF-CC7 complex
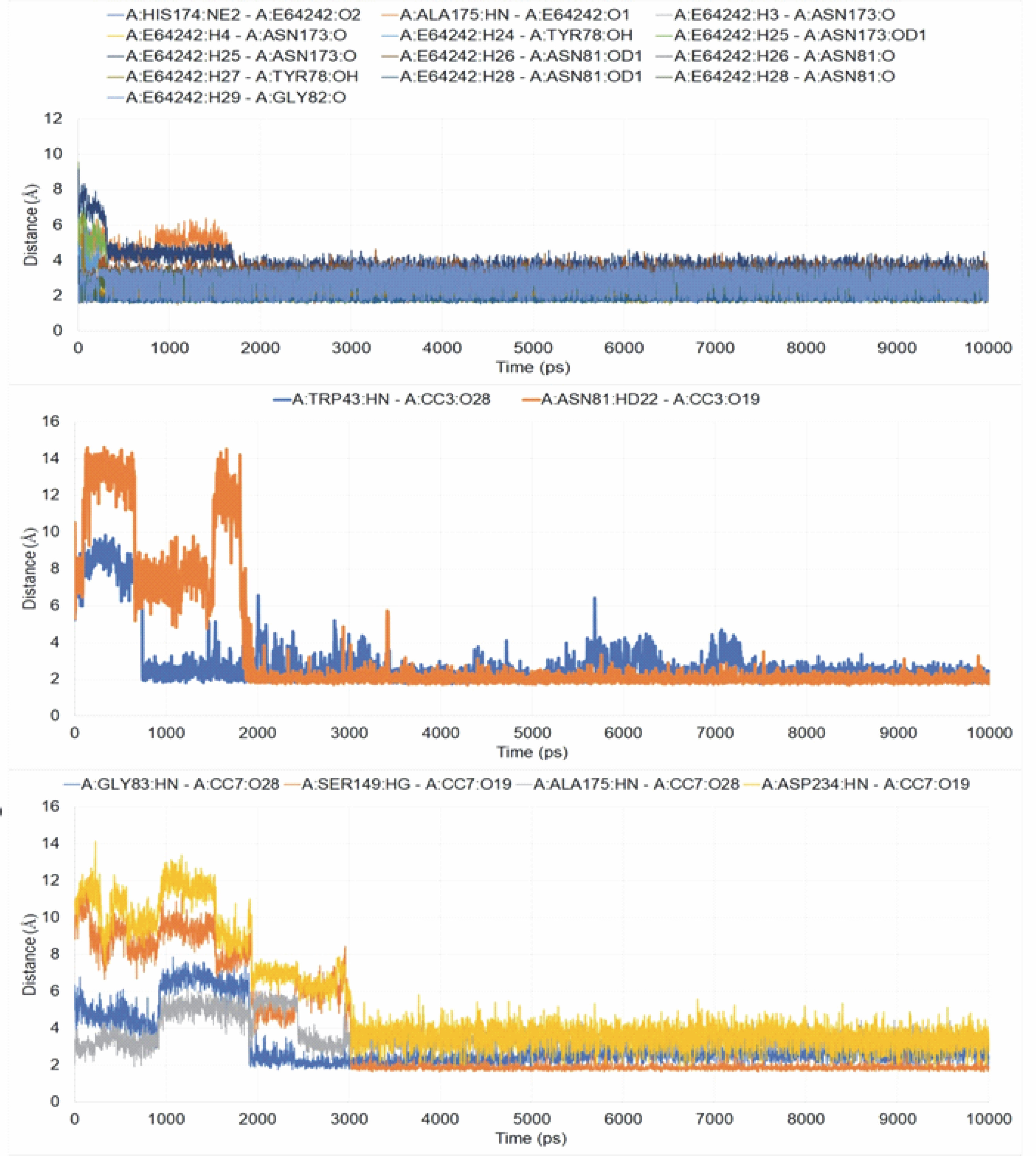
The interactions of the average protein-ligand complexes obtained after the MD simulation study showed that the co-crystal ligand formed 13 hydrogen bonds with the protein 3BPF, whereas CC3 formed only two and CC7 formed only four hydrogen bonds with 3BPF. Other than the hydrogen bonds, all the compounds formed some hydrophobic interactions (Fig. 7) in the active binding site of 3BPF. Amino acid residues such as Trp 43, Asn 81, Gly 82, Gly83, Leu 84, Ser 149, Asn 173, Ala 175, Thr 233, and Asp 234 were involved in the interactions with trioxane molecules. Later, the fluctuations of the distances of hydrogen bonds formed were calculated, and we found that for E64, all the hydrogen bonds were stabilized within 2 ns and the distances were maintained under 5 Å after 2 ns. Even for CC3, the hydrogen bonds were stabilized within 2 ns, and the distances were maintained under 5 Å after 2 ns of the simulation period. For CC7, the hydrogen bonds were highly fluctuated within 3 ns, but subsequently, they were stabilized and fluctuated under 5 Å until the end of the simulation period (Fig. 8).

MMPBSA-based binding free energy
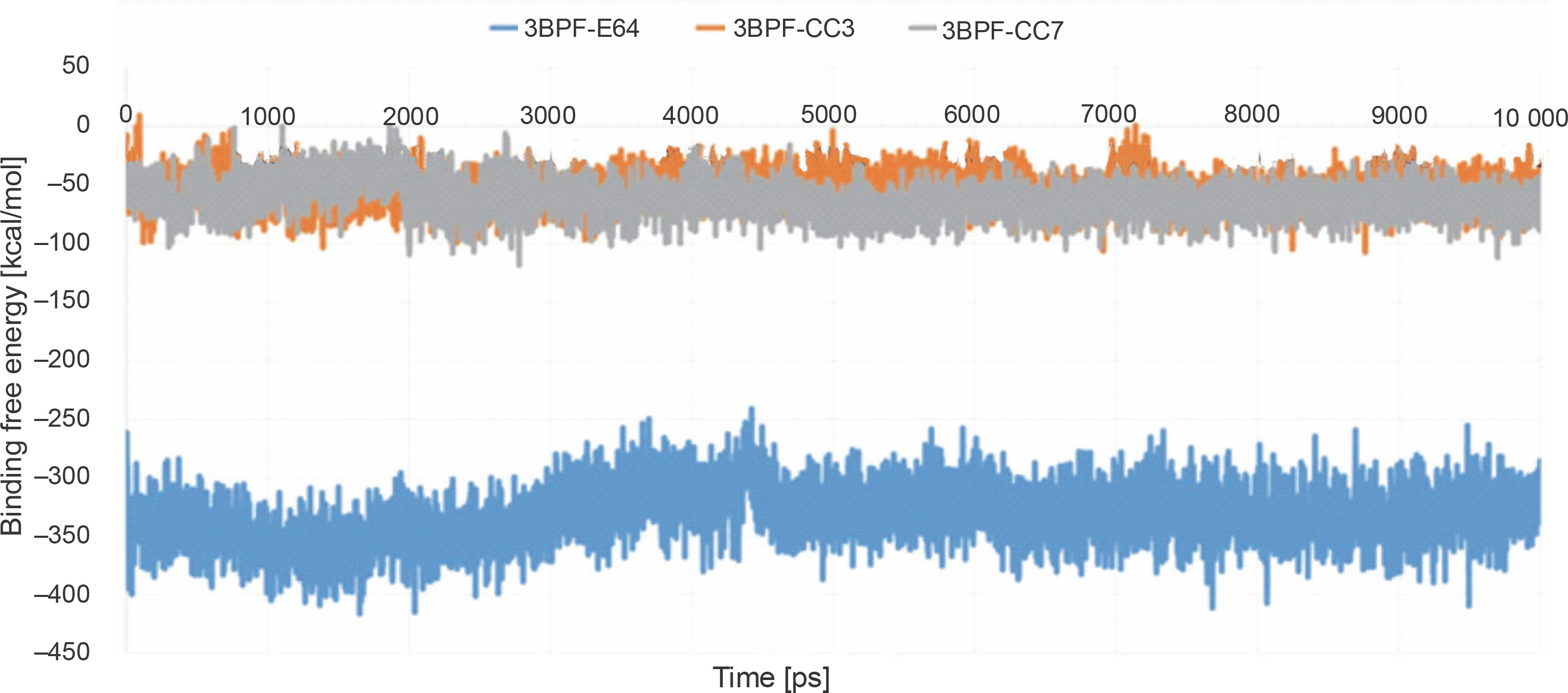
After completing the MD simulation, the MM-PBSA-based binding free energies were calculated for all the generated conformations, and the average binding free energy was then calculated for each protein-ligand complex (Fig. 9). The binding free energy of the test compounds (CC3: –55.078 kcal/mol and CC7: –62.241 kcal/mol) were found to be considerably higher than that of the co-crystal ligand E64 (–33.079 kcal/mol). This indicated that the test compounds (CC3 and CC7) formed thermodynamically stable complexes with the target protein, which are comparable to that of the complex formed with the co-crystal ligand E64.
Docking for selectivity study
Table 6 presents the results of docking (LibDock scores) for the selectivity study of compounds CC3 and CC7 against Cat K and Cat L. Both CC3 and CC7 exhibited selectivity against both the host proteins in terms of LibDock scores. Compound CC3 showed higher selectivity on Cat K with the LibDock score of 140.913, whereas compound CC7 exhibited higher selectivity on Cat L with the LibDock score of 130.701. Selectivity of trioxane compounds (CC3 and CC7) was comparable to that of the co-crystal ligands (O96 and I0X). Figure 10 and Figure 11 shows binding modes and interactions (2D and 3D) of docking for compounds CC3 and CC7 against the host homologous cysteine proteases Cat K and Cat L, respectively. For human proteases, the interactions between protein amino acid residues and ligand atoms were observed with the formation of hydrogen and hydrophobic bonds. For Cat K, two H-bonds were formed with both the compounds CC3 and CC7, while, for Cat L, two and three H-bonds were observed for compounds CC3 and CC7, respectively. The co-crystal ligand formed six and five H-bonds for Cat K and Cat L, respectively. Amino acid residues such as Gln 21, Gly 23, Gly 69, His 164, Ala 163, and Gly 165 of Cat K and Cat L proteins were involved in H-bond formation with the ligand molecules CC3 and CC7.
Fig. 10
Binding modes and docking interactions diagrams of compounds CC3 (A), CC7 (B), and the co-crystal ligand O96 (C) (left: 2D interaction, right: 3D interaction) against cathepsin K (3OVZ); dotted green lines indicate conventional H-bonding interactions, and dotted yellow and purple lines indicate different types of hydrophobic interactions
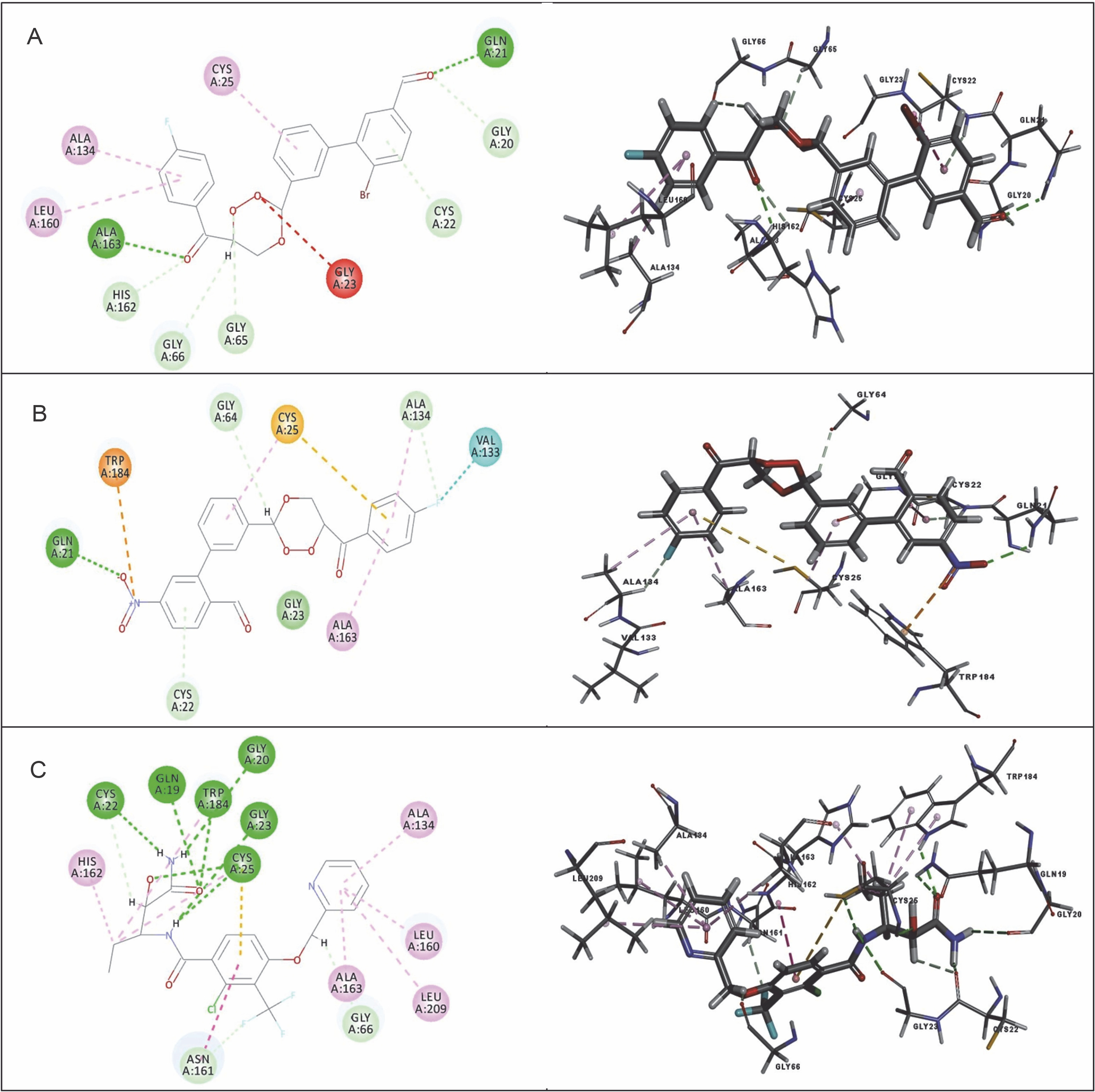
Table 6
LibDock scores of 1,2,4-trioxane derivatives CC3 and CC7 against cathepsin K and cathepsin L
| Compounds code | LibDock score | Number of H-bonds | ||
|---|---|---|---|---|
| 3OVZ | 3OF9 | 3OVZ | 3OF9 | |
| CC3 | 140.913 | 120.04 | 2 | 2 |
| CC7 | 137.396 | 130.701 | 2 | 3 |
| Co-crystal inhibitor | 134.625 | 152.045 | 6 | 5 |
Fig. 11
Binding modes and docking interaction diagrams of compounds CC3 (A), CC7 (B), and the co-crystal ligand I0X (C) (left: 2D interaction, right: 3D interaction) against cathepsin K (3OF9); dotted green lines indicate conventional H-bonding interactions, and dotted yellow and purple lines indicate different types of hydrophobic interactions
Discussion
In our study, 15 new 1,2,4-trioxane derivatives were designed by the molecular manipulation approach of rational drug design (RDD) and reported as novel FP-2 inhibitors with predictive (in silico ) antimalarial potential against P. falciparum. In ART-based antimalarial drugs, 1,2,4-trioxane is believed to be the key pharmacophoric moiety responsible for their antiparasitic effect (Rudrapal et al., 2017). In view of this fact, novel 1,2,4-trioxane derivatives were designed by structural modification of 1,2,4-trioxane pharmacophoric skeleton with the incorporation of substituted bulky aryl moieties. Rudrapal et al. (2017) designed a series of 1,2,4-trioxane derivatives with different alkyl/aryl/heteroaryl substitutions at the C-3 position of the 1,2,4-trioxane scaffold. They reported that the presence of aryl/heteroaryl moieties could greatly modulate the antimalarial efficacy of the basic trioxane scaffold. Therefore, no alternations were made in the parent structural framework of 1,2,4-trioxane skeleton which is an essential requirement for the antimalarial activity, but a structural modification was made by incorporating biologically/pharmacokinetically relevant bulky aryl substituents (4-fluorobenzoyl and substituted biphenyl-2-carbaldehyde). Molecular docking is a virtual tool intended to find the best binding orientation of small molecules bound to their target protein molecules. It is used to predict the binding affinity and biological efficacy of small molecules (Ghorab et al., 2020; Abdel-Hamid et al., 2014). Moreover, docking plays an important role in the identification of bioactive molecules based on the target protein structure in RDD and discovery program (Rudrapal et al., 2018). The docking protocol was validated by redocking the co-crystal ligand into the predicted binding sites of the target receptor protein followed by analyzing the binding modes of the test compound in the docked complex by comparing with the binding modes of the cocrystal inhibitor E64 (Rudrapal et al., 2019). The cocrystallized ligand was re-docked using flexible docking simulations into the original structure of the receptor molecule by setting all the docking parameters to the software’s default values. The validation of the docking method confirmed the accuracy of the procedure with an acceptable RMSD of less than 2 Å. The results of our docking study are consistent with previous results reported in the literature. Kalita et al. (2019) recorded similar observations while performing validation of the docking method using the FP-2 protein (3BPF) in their study. This further substantiates the validation of the docking method reported in our study.
The protein-ligand docking was performed to predict the binding affinity of the test compounds CC1–CC15 as possible novel antimalarial agents with inhibitory activity against P. falciparum FP-2. FP-2 is a cysteine protease enzyme that plays an important role during the process of hemoglobin degradation in the food vacuole of P. falciparum during the erythrocytic life cycle of the parasite (Rudrapal et al., 2017; Grazioso et al., 2012). In the protein-ligand docking study, the LibDock program successfully docked all the compounds CC1–CC15 into the binding pocket of protein molecules. The compounds could bind well with the predefined active site residues of the predicted receptor spheres depicted in the receptor grid model. Higher binding affinities in terms of LibDock scores were observed for all the deigned compounds as compared to the co-crystal inhibitor E64. In the protein-ligand interaction analysis, it was found that the basic 1,2,4-trioxane moiety interacted with the amino acid residues (such as Gln 36, Ser 41, Cys 42, and His 174 depicted in the results section) at the active site predominantly through the formation of H-bonds, and aryl rings and substituent groups such as ‒OH, ‒CH3, and –NO2 were also involved in interaction by H-bonding and various hydrophobic interactions such as Van der Waals and pi-alkyl bonds. A strong interaction of the trioxane pharmacophore with the predicted binding sites of FP-2 was observed. Substituents such as –OH and –CH3 further modulated the strength of interaction to achieve optimal interaction. In a previous study, Rudrapal et al. (2018) reported that 1,2,4-trioxane interacted with the FP-2 protein through the formation of hydrogen and hydrophobic bonds. In their study, besides the trioxane pharmacophore, various substituents/moieties also contributed to the molecular interactions between the key pharmacophoric trioxane structure and various amino acid residues of the FP-2 protein. According to their reports, the trioxane pharmacophore significantly interacted with certain key amino acid residues such as Cys 42, Gly 83, and His 174, with the LibDock score ranging from 93 to 126 against FP-2. Although our present study is consistent with previous findings, the trioxane derivatives reported here possess greater FP-2 inhibitory potential (in terms of LibDock score and H-bonding interactions) than trioxanes reported by Rudrapal et al. (2017). The present trioxane derivatives exhibited comparatively higher LibDock scores (>100) with the formation of more number of H-bonds. This clearly reflects the stronger binding affinity/inhibitory potential against the FP-2 protein. From receptor-ligand interactions, it is also evident that the substituted aryl moieties (HC=O, C=O, and NO2) further modulate the binding affinity besides the direct inhibitory role of the trioxane scaffold against FP-2. Furthermore, literature also confirm that the inhibition of the FP-2 protein by structural scaffolds (such as 1,4-benzodiazepine) other than trioxane is due to the involvement of the interacting amino acid residues such as Cys 42 and His 174 (Giovanni et al., 2012). These observations further strengthen our findings. Considering all these, it is apparent that the 1,2,4-trioxane scaffold- based derivatives designed in our study could offer a novel pharmacophoric moiety for further modification toward the development of new trioxane-based compounds/agents as promising P. falciparum FP-2 inhibitors with in vitro/in vivo antimalarial activities. However, subtle structural modifications must be performed to improve the antimalarial potency and to avoid undesired toxic effects such as hepatotoxicity and mutagenic properties.
In our study, all compounds obeyed “Lipinski’s rule of five” and “Veber rule.” According to Lipinski’s rule, compounds are more likely to be drug-like and orally bioavailable if they obey the following criteria: LogPo/w (octanol/water partition coefficient) ≤ 5, MW ≤ 500, nHBAs ≤ 10, and nHBDs ≤ 5 (Jain et al., 2008). To further substantiate drug-likeness, Veber et al. (2002) stated that compounds with ≤10 RotB and molecular PSA of ≤140 A2 are more likely to show optimum membrane permeability and good bioavailability. In ADMET prediction, aqueous solubility, BBB penetration, CYP P450 2D6 inhibition, hepatotoxicity, intestinal absorption, and plasma protein binding were in acceptable limits (Rudrapal et al., 2017; Ponnan et al., 2013; Rudrapal et al., 2016b). The predicted toxicity data such as LD50 in mice and rats, Ames mutagenicity, DTP, and skin irritancy did not cross the limit of toxicity. Previous studies support the results of our present studies related to the prediction of toxicity parameters of trioxane derivatives (Rudrapal et al., 2019). Thus, it can be stated that the designed trioxane derivatives possess drug-likeness and ADMET properties within the acceptable range and do not have any predictive toxic features.
MD simulation study, which provides more accurate and reliable information than simple molecular docking study, was performed to observe the stability of the protein-ligand complexes. In the MD simulation analysis, both protein and ligand structures remain flexible, thus allowing for an induced fit into the binding site of the receptor around the introduced ligand (Lin et al., 2011). MD simulation was analyzed on the basis of RMSD, RMSF, and ROG as a function of time. Fluctuations of the distances of the hydrogen bonds within the simulation period of 10 ns were determined. The MM-PBSA method is a popular approach to estimate the free energy of the binding of small ligands to proteins. These methods are typically based on MD simulations of the receptor-ligand complex and are therefore intermediate in both accuracy and computational effort between empirical scoring and strict alchemical perturbation methods (Rastelli et al., 2010).The MM-PBSA-based calculation of binding free energy (ΔG) is one of the important parameters to estimate the binding affinity of a compound to a protein target as well as the thermodynamic stability of the protein-ligand complex (Genheden and Ryde, 2015). This technique provides a fast and accurate prediction of absolute binding affinity of a compound within the active binding site of a target protein in the form of binding free energy, which is very important for stability and particular potency of the compound (Rastelli et al., 2010). Analysis of the results of MD simulation revealed that the trioxane compounds formed stale complexes with the Plasmodium cysteine protease FP-2. The fluctuations of the amino acid residues in all the three cases of RMSD, RMSF, and ROG determinations were very small, and no significant differences were observed. The MD simulation study thus confirms the predictivity of binding affinity of the trioxane compounds with the FP-2 molecule. Furthermore, the analysis of MM-PBSA-based binding free energies revealed the formation of thermodynamically stable (during the simulation period of 10 ns) complexes between trioxanes and the FP-2 molecule. From the above observations, it is apparent that the trioxane pharmacophore along with the structural substituents contributed significantly to the formation of strong and stable protein-ligand complexes. Musyoka et al. (2016b) demonstrated well-defined and stable protein-ligand complexes where nonpeptidic compounds (chalcones and isoquinolenes) interacted with the involvement of some key amino acid residues (Asn, Cys, Gln, and His) from Plasmodium FP-2 protease, which substantiates the FP-2 inhibitory potential of trioxanes reported in this study.
Results of the selectivity study indicate that in addition to higher binding affinities against Plasmodium cysteine protease FP-2, the trioxane compounds showed lesser selectivity on human homologous proteases Cat K and Cat L. This implies that the trioxane compounds have more binding affinity for the parasitic protease (FP-2) over the host proteases (Cat K or Cat L). The enzymatic selectivity for the human proteases is not significant for the trioxane molecules. This study therefore proves that trioxane molecules are devoid of any interactions/toxic effects on humans. In the docking study against the FP-2 molecule, two best scored compounds CC3 and CC7 with well-defined H-bonding interactions with the FP-2 protein formed 7 and 5 H-bonds, respectively. In human Cat K and Cat L, the number of H-bonds between amino acids and ligands was comparatively lower to that of Plasmodium FP-2. The molecular interactions predicted in docking in terms of H-bonding occupancy and bond strength were comparatively less significant for the human proteases cathepsins than for Plasmodium protease FP-2. Similar finding have been observed in recent studies reported in literature (Musyoka et al., 2016a; Musyoka et al., 2016b), which substantiates the results of our study. The authors reported marginal selectivity of their designed FP-2 inhibitors on human homologous cathepsins.
Conclusions
In the present study, 15 new trioxane derivatives were designed based on the 1,2,4-troxane pharmacophoric scaffold of artemisinin by the molecular modification approach. The designed compounds were investigated by molecular modeling studies, including molecular docking, drug-likeness, ADMET, and toxicity assessment and further MD simulation and MM-PBSA studies. Among the 15 trixoane derivatives, two compounds, namely CC3 and CC7, were found to exhibit the highest binding affinity against the P. falciparum cysteine protease FP-2 enzyme in terms of LibDock score. These compounds are therefore suggested to be potent molecules with FP-2 inhibitory activity. All compounds exhibited satisfactory in silico drug-likeness, ADMET, and toxicity properties. No compounds violated the Lipinski’s rule of five, and the drug-likeness scores of all the compounds were within the acceptable range. MD studies further confirmed the antimalarial potential of the compounds CC3 and CC7 with the formation of well-defined and stable receptor-ligand interactions against the P. falciparum FP-2 enzyme. The MM-PBSA study indicated that compounds CC3 and CC7 formed thermodynamically stable complexes with the target protein FP-2, which are comparable to the complexes formed with the co-crystal ligand E64. Furthermore, in addition to the inhibitory activity of CC3 and CC7 against Plasmodium cysteine protease FP-2, the selectivity on human cathepsins (Cat K and Cat L) was identified. It can be concluded that the newly designed 1,2,4-trioxane derivatives can be further investigated for in vitro and in vivo antimalarial activities for their possible development as potent antimalarial agents effective against resistant strains of P. falciparum.