






Introduction
Chondromyxoid fibromas (CMF) are rare benign bone lesions representing 0.5–1% of all bone tumours [1]. Chondromyxoid fibromas is characterised by a moderate local recurrence rate and a small risk of malignant transformation [1]. Histologically, CMF is composed of variable amounts of chondroid, myxoid, and myofibroblastic elements [2], and it usually arises in the long bones of the lower limbs, typically affecting the metaphysis (88%) [3]. It most commonly occurs between the second and third decades of life [4], and more frequently in males [5].
The chondromyxoid fibromas was recognised as a distinct pathologic entity in 1948 by Jaffe and Lichtenstein, who provided its first histological description [6]. However, it still presents a serious clinical challenge, and in most cases is diagnosed mainly by exclusion (per exclusion). Here, we report the case of a 66-year-old female who, due to a non-specific radiological and histological presentation of CMF, had been subjected to a lengthy and wearing process of subsequent faulty diagnostic steps. The case emphasises the importance of genetic studies and a multidisciplinary approach to be implemented at the initial stages of the diagnosis of CMF.
Case report
A 66-year-old female was admitted to the Department of Neurosurgery in August 2021 with a tumour in the area of the right fibula. An axial and sagittal T1- and T2-weighted magnetic resonance imaging (MRI) without contrast enhancement consisting of proton density turbo spin-echo sequence of the right lower limb, performed on admission, showed a 18 × 8 × 13 mm, well-defined, irregular, and polycyclic hyperintense lesion exhibiting a homogenous signal (Fig. 1 A). The tumour was compressing the anterior cortical layer of the fibula. The location and the radiological image of the lesion were suggestive of a neuroma of the right tibial nerve, and the patient was referred to excision surgery. The completion of the procedure (R0 resection) revealed that, contrary to the initial diagnosis, the tumour originated not from the nerve but from the bone below the head of the right fibula. Given the absence of any direct connection between the tumour and the nerve, it was suspected post- operatively that the lesion could be a chondroma. However, this remained only a preliminary hypothesis, and the tissue was sent for further pathological examination to confirm the definitive diagnosis.
Fig. 1
Radiological and histopathological presentation of the lesion. Radiographic (proton density – weighted magnetic resonance imaging sequence) study of the lesion (white arrow) in right lower limb (A), histopathological analysis of the neoplasm. Haematoxylin and eosin staining showing mean cellularity and production of osteoid foci (black arrow) (20× magnification) (B), immunohistochemical staining demonstrates negativity for S-100 marker (C) and low Ki-67 index (D), (20× magnificationFig. 1. Radiological and histopathological presentation of the lesion. Radiographic (proton density – weighted magnetic resonance imaging sequence) study of the lesion (white arrow) in right lower limb (A), histopathological analysis of the neoplasm. Haematoxylin and eosin staining showing mean cellularity and production of osteoid foci (black arrow) (20× magnification) (B), immunohistochemical staining demonstrates negativity for S-100 marker (C) and low Ki-67 index (D), (20× magnification
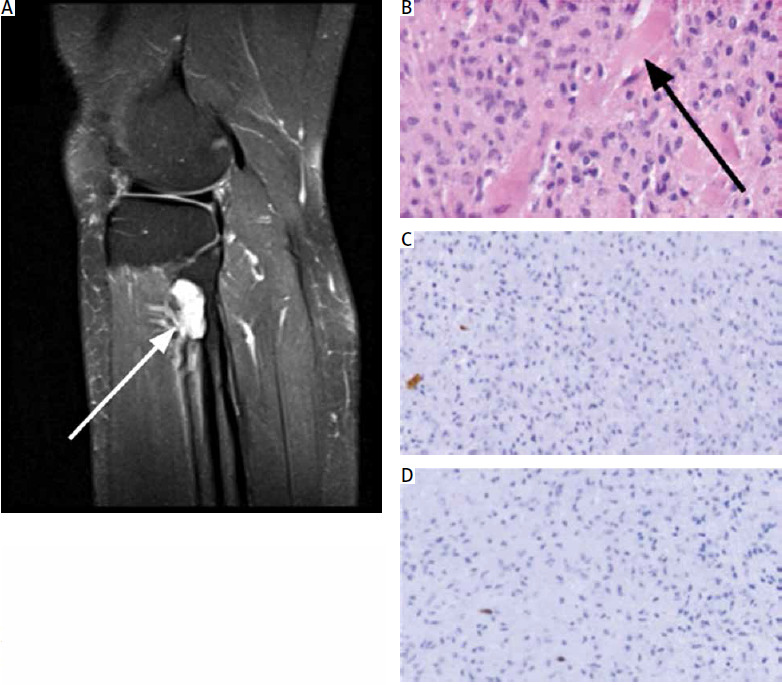
Histological analysis of the specimen showed that the lesion consisted of cells producing osteoid foci of moderate cellularity, rich in mononuclear cells: small, round or fusiform, with mild atypia. Mitotic figures and necrosis were absent (Fig. 1 B). Immunohistochemical staining showed negativity for S100 protein (a classical neuronal marker), and low Ki-67 (9–10%) index (Figs. 1 C, D). Due to osteoid production, the second diagnosis of a low-grade parosteal osteosarcoma, prognostically much less favourable for the patient, was put forward.
Due to ambiguous histological findings, such as a low Ki-67 index, and incongruent MRI results (notably the absence of bone infiltration), the tissue samples were forwarded to the National Institute of Oncology in Warsaw. This referral was made for further immunohistochemical staining procedures and an in-depth examination of alterations in the GNAS gene. Activating mutations of this gene at codon R201 (exon 8) and, more rarely, codon 227 (exon 9) are characteristic of fibrous dysplasia of bone, which may histologically mimic osteosarcoma [7]. Using direct Sanger sequencing, no mutations were found in any of the examined gene segments, thus ruling out fibrous dysplasia.
An extended panel of immunohistochemical markers included smooth muscle actin (SMA) and markers of a low-grade osteosarcoma, i.e. MDM2 [8] and RB1 [9]. The findings for S-100 and Ki-67 aligned with initial observations. The tumour demonstrated negativity for MDM-2 and positivity for RB-1 and SMA, thereby strengthening the exclusion of both fibrous dysplasia and osteosarcoma, which was in agreement with genetic analyses.
In summary, a comprehensive approach involving radiological, histological, and genetic analyses pointed towards a chondromyxoid fibroma, leading to its confirmation as the definitive diagnosis. Consequently, the diagnostic journey concluded favourably for the patient, identifying the lesion as benign and not necessitating systemic therapies. The patient is undergoing monitoring through MRI at 6-month intervals and presently maintains a good condition.
Discussion
In this report, an unusual case of CMF has been presented, which, over the course of its diagnosis, has been mistaken for 3 different entities: a neuroma, a chondroma, and an osteosarcoma. Chondromyxoid fibroma has been recognised for its potential to mimic other lesions on MRI, which has often led to its misdiagnosis as either benign [10–12] or malignant growths [13]. In this case, a polycyclic hyperintense change that was initially interpreted on MRI has been classified as a neuroma. Histopathologically, CMF has typically been characterised by lobulated areas containing chondroid and myxoid elements, with cellularity being low in the centre and higher at the periphery, where osteoclastic giant cells may also be found. Features such as hyaline cartilage, mitotic figures, and necrosis are uncommon findings and have not been observed in the microscopic images of the reported case [14]. Additionally, the sample has mimicked “osteoid production”, which has altogether been highly suggestive of parosteal osteosarcoma [15].
The role of immunohistochemistry in diagnosing CMF remains unclear [16]. Studies by Söder et al. [17] and Sreedharanunni et al. [14] have shown positivity for S-100, contrasting with our case where the tumour showed no immunoreactivity for S-100. Additionally, our results did not align with the high proliferation index indicated by Ki-67 as reported by Veras et al. [18].
The World Health Organisation classification states that molecular analysis is not required for diagnosing CMF fibroma [5]. However, in our case, an assessment of the GNAS gene was undertaken to exclude fibrous dysplasia. The identification of alterations in the GRM1 gene is considered highly specific and definitive for CMF [5, 19]. The dysregulation of the glutamate receptor (GRM1), involving chromoplexy and chromothripsis, leads to the recombination of GRM1 and the upregulated expression of its transcripts, which is believed to contribute to disease progression. These genetic changes form the basis for differentiating CMF, as GRM1 expression in other cartilaginous origin neoplasms is either absent or very low. Nevertheless, it is estimated that about 10% of CMFs do not demonstrate upregulation of GRM1 [1, 5]. In the presented case, mutations in the GRM1 gene were not tested. Although this might be perceived as diagnostic negligence, the adopted measures were sufficient to allow (through a process of exclusion) us to satisfactorily confirm the diagnosis.
Conclusions
The presented case highlights the diverse methods utilised in the diagnosis of chondromyxoid fibroma, showcasing an unusual clinical presentation of the lesion which, mimicking a neuroma radiologically and a low-grade osteosarcoma pathologically, was finally revealed to be a benign tumour with a favourable prognosis. Documenting such rare cases enriches the medical literature on this topic and potentially improves the diagnostic process. This is important not only for the implementation of adequate therapeutic strategies but also for the psychological well-being of the patient. It is our hope that this report will serve healthcare professionals as a valuable reference during the assessment and diagnosis of similar cases.