






Introduction
Bile duct obstruction leads to biliary atresia (BA), a serious liver disorder that develops into liver fibrosis [1]. Biliary atresia, known as “neonatal hepatitis”, is the most prevalent cause of pediatrics liver transplantation worldwide; also, it is a significant contributor to infant cholestasis [1]. It has been challenging to find novel treatments that would reduce the requirement for surgery due to the complexity of the problem [2]. Biliary atresia is a fatal condition that causes obstruction of the bile duct which can strike infants [3]. Over one third (37%) of the various etiologies of newborn cholestasis in Egyptian patients can be attributed to BA [4]. Biliary atresia is caused by an autoimmune and fibrosing obstruction of the extrahepatic bile ducts [5].
At the time of diagnosis, the extrahepatic bile ducts are completely clogged. At the tissue level, there is significant fibrosis, partial or total loss of the epithelial lining of the extrahepatic bile ducts, and occasional areas of inflammation [6]. Contrarily, intrahepatic bile ducts are frequently hyperplastic, embedded in portal tracts that exhibit variable degrees of fibrosis and inflammation, and encircled by lobules that exhibit cholestasis-like traits and enormous multinucleated hepatocytes [6].
Biliary atresia is difficult to diagnose due to the disease’s overlapping clinical and imaging symptoms [7]. Non-hepatic congenital abnormalities and earlier-onset jaundice, which are frequently present at birth, afflict about 10% of affected infants [8]. Congenital, fetal, and prenatal BA are the other names for this disease. Splenic abnormalities can be present alone or conjunction with one or more other issues in a variant known as biliary atresia splenic malformation (BASM) syndrome, which affects 8-12% of babies with the embryonic form of the condition [9].
Infants with BASM have a stronger correlation with maternal diabetes mellitus, called syndromic type of BA, and may not respond well to hepato-portoenterostomy [1]. The surgical course of treatment for BA is known as Kasai portoenterostomy (KPE), which is carried out surgically within the first 60 days of life to enhance the prognosis [10]. There are several etiologies; some are syndromic while others are viral in origin. Biliary atresia is based on unclear exact etiologies [11]. Factors associated with genetic predisposition are proposed, but they have not yet been fully proven [12].
Beta secretase and γ secretase break the precursor protein for amyloid β (Aβ) to produce the Ab1-40 or Ab1-42 Aβ peptide, which has 40 or 42 amino acids, respectively. The N terminal of the peptide is produced by β secretase using Aβ precursor proteins, and the C terminal is produced by γ secretase. The major pathogenic peptide, Ab1-42, is extremely hydrophobic and has a propensity to group together into oligomers and fibrils [13].
Because of the additional two amino acids’ increased propensity to misfold and subsequently clump, Ab1-42 is thought to be more neurotoxic [14]. Around the bile ducts in the livers of BA patients, Aβ deposition was found. Amyloid β peptide is produced proteolytically by processing of a transmembrane protein, amyloid precursor protein (APP), by β- and γ-secretases [9]. A novel pathogenic mechanism has been presented [15]. Cholangitis, portal hypertension, and variceal hemorrhage are the most frequent side effects of BA that necessitate liver transplantation [16, 17]. The Aβ deposition in the livers of BA babies reduced liver regeneration via impairing mitochondrial respiration and mammalian target of rapamycin (mTOR) signaling [18]. So, the goal of this work was to assess the presence of amyloid β precursor protein (APP) gene expression in BA patients in comparison with other causes of neonatal cholestasis. This could be used to explore the role of Aβ in the pathophysiology and diagnosis of neonates with BA.
Material and methods
Study design and patient enrolment
Patients were studied from March 2022 to December 2022. They attended the outpatient clinic of the Paediatric Hepatology, Gastroenterology, and Nutrition department at the National Liver Institute, Menoufia University, Shebin El Kom, Menoufia, Egypt.
Patients’ inclusion and exclusion criteria
All patients were identified using the inclusion and exclusion criteria listed below.
Inclusion criteria included new-born with a diagnosis of BA. Patients with BA had a BA score when they were diagnosed. The intraoperative cholangiogram added further confirmation to the diagnosis [19]. Control participants were diagnosed with metabolic associated fatty liver diseases (MAFLD) by the pathological findings in liver biopsy.
Pathological diagnosis
Using the Ishak score, the degree of liver fibrosis was determined. The Ishak score was found to offer the best predictive validity for an early liver transplant in BA patients as well as the lowest interobserver variability. The Ishak score is advised in everyday clinical practice for assessing liver biopsies in BA patients due to its straightforward applicability [20].
Exclusion criteria included any case with a diagnosis of neglected BA, successful and failed post-Kasai, galactosemia, tyrosinemia, inspissated bile syndrome and sepsis-induced cholestasis.
Examination measurements
Clinical information (such as age, sex, and complaint), a family history of similar illnesses, laboratory data, and ultrasonographic data were all obtained from the medical records.
Complete blood count (CBC) tests were performed in the laboratory using the Sysmex XT 1800i (Sysmex Corporation, Kobe, Japan) system to measure hemoglobin (Hb), white blood cells (WBCs), platelets, prothrombin time, and international randomized ratio (INR), as well as automated hemostasis testing with the Sysmex CS-1600. Total bilirubin (TB) and direct bilirubin (DB), γ-glutamyl transferase (GGT), aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase (ALP), serum albumin, and TB were all measured using the Cobas 6000 analyzer (c501 module) (Roche Diagnostics GmbH, D-68305 Mannheim, Germany). All the infants under study had their Aβ precursor protein gene expression analyzed using qPCR. Ultrasound with Doppler (Toshiba, Japan) of the abdomen was performed, laying particular stress on liver architecture and echogenicity, liver size and spleen size. Each patient’s liver was biopsied utilizing complete core biopsy needles and automated cutting-type needles. The percutaneous method was carried out with a 16 G needle, and the length of the sample was at least 20 mm [21].
Tissue homogenization
The tissue was extensively homogenized using the Tissue Ruptor II (Qiagen, Hilden, Germany). The mixture was then centrifuged for 20 minutes at 4000 rpm. The cell supernatant was then lastly gathered and used to extract RNA.
Total RNA extraction
After homogenizing liver tissue, the RNeasy Blood/Tissue Mini kit from Qiagen (Hilden, Germany) was used to extract and purify RNA. As directed by the manufacturer, the operation was carried out.
cDNA synthesis (reverse transcription)
The QuantiTect Reverse Transcription Kit (cat. no. 205310, Qiagen, Hilden, Germany) was used to complete the reverse transcription process. All of the components necessary for first-strand cDNA synthesis were included in the reverse-transcription master mix, which was made in a 20 L total volume on ice. In order to inactivate Quantiscript Reverse Transcriptase, the reaction mixture was incubated for 15 min at 42°C then for 3 min at 95°C. After putting the reverse-transcription reactions on ice, real-time PCR started immediately.
APP gene amplification by qPCR
The AAP gene expression level was amplified from mRNA using Amyloid beta (A4) Precursor Protein QuantiTect Primer Assay [Hs_APP_1, QuantiTect Primer Assay, Gene Globe ID: QT00050554] cat no: 249900 (Qiagen, Germany) and QuantiTect SYBR Green PCR Kit cat no: 204141 (Qiagen, Germany) and Hs_ACTB_1_SG QuantiTect Primer Assay (β-actin) cat no: 249900, ID: QT00095431 as a housekeeper gene. All samples were analyzed using the 5 plex Rotor Gene PCR Analyzer (Qiagen, Germany).
The real-time cycler was programmed as: activation step for 15 minutes at 95°C for Hot-Star Taq DNA Polymerase activation. Three-step cycling: denaturation for 15 s at 94°C, annealing for 30 s at 55°C, extension for 30 s at 70°C, for 40 cycles. Moreover, the expression levels were normalized to β-actin levels as a reference gene. The relative gene expression levels (fold change) for APP normalized to an internal control (β-actin) and relative to a calibrator (negative control sample) were calculated using the equation 2-∆∆Ct test/control.
Ethics approval and consent to participate
All participants’ legal guardians signed informed consent after receiving a brief and detailed description of the study’s goals. Also, the Menoufia Faculty of Medicine’s local ethical scientific committee gave approval to the current study plan (Registration number: NLI IRB 00003413 FWA0000227).
Sample size calculation
Sample size was calculated using PASS 11.0, based on the previous trial by Tian et al. [3] both Aβ mRNA and protein significantly increased in livers of infants with BA. Amyloid β was elevated significantly in patients with BA and positively correlated with liver injury progression. Assuming a standard deviation of 5 (standard value of 1.96), using the study power test of 80% with a drop-out rate of 10%, a sample size of 35 infants was required (16 infants with BA, 14 infants with neonatal cholestasis and 5 other healthy infants as the control group).
Outcomes of the study
Assessment of the level of APP gene expression among children with (BA, neonatal cholestasis [NC], and control groups) and its function in pathogenesis and diagnosis of BA infants.
Evaluation of the association of APP gene expression with GGT, ALP, AST, and TB.
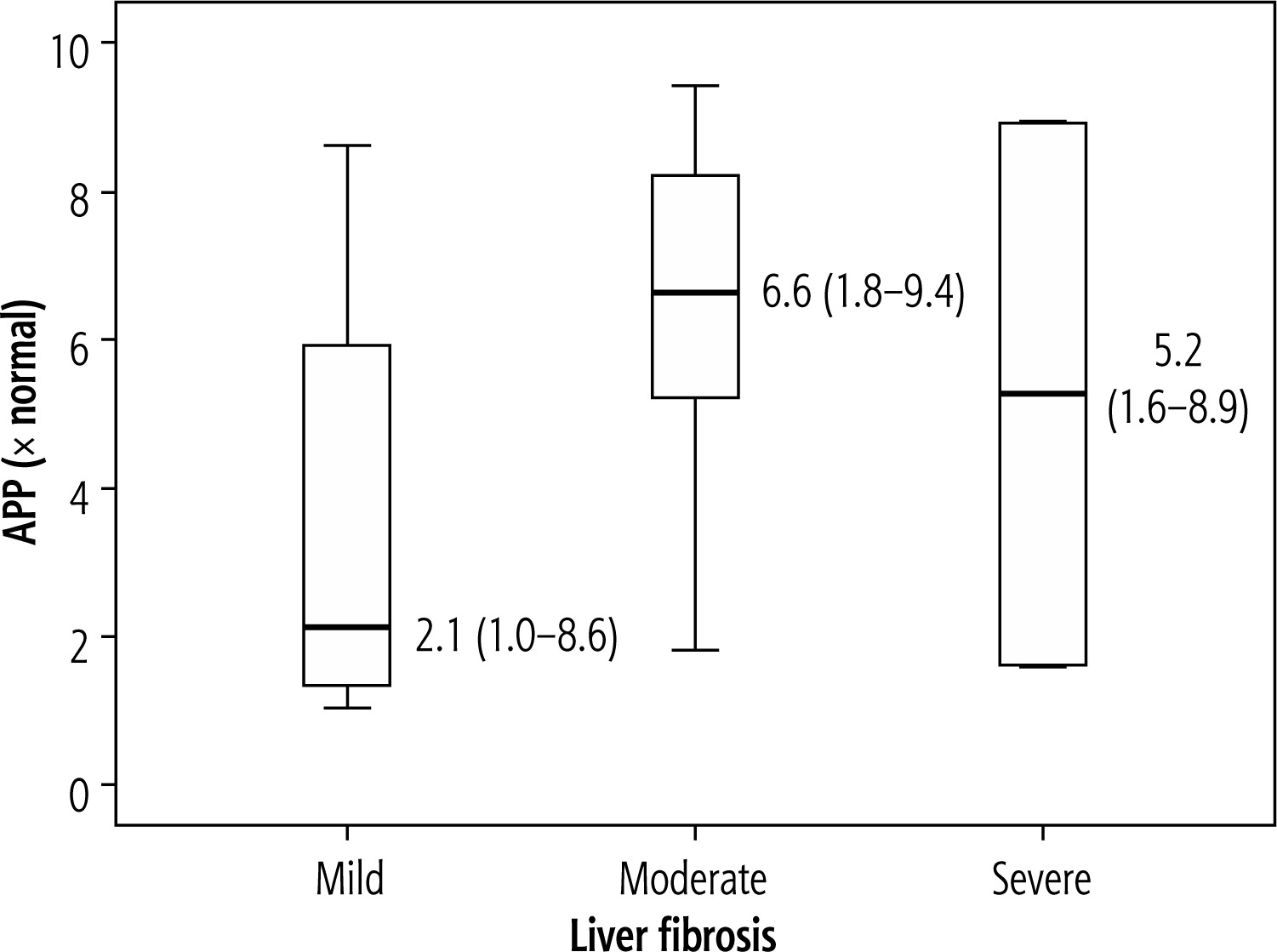
Assessment of the relationship between the level of APP gene expression and the severity of liver fibrosis.
Statistical analysis
The results were tabulated and statistically analyzed using SPSS V.25 (IBM Corporation, 1 Orchard Rd, Armonk, NY 10504, USA) and Microsoft Excel 2019 (Microsoft Corporation, One Microsoft Way Redmond, WA 98052-6399, USA). The descriptive statistics included mean (x), median, and SD. The Kolmogorov-Smirnov test was used to determine whether the variable distribution was normal. Group comparisons for categorical variables were assessed using the χ2 test (Fisher exact). The Mann-Whitney U test and the Kruskal-Wallis test were used to test quantitative variables with non-normal distributions. The Spearman coefficient was used to ascertain the correlation between quantitative variables. P ≤ 0.05 was selected as the level of statistical significance.
Results
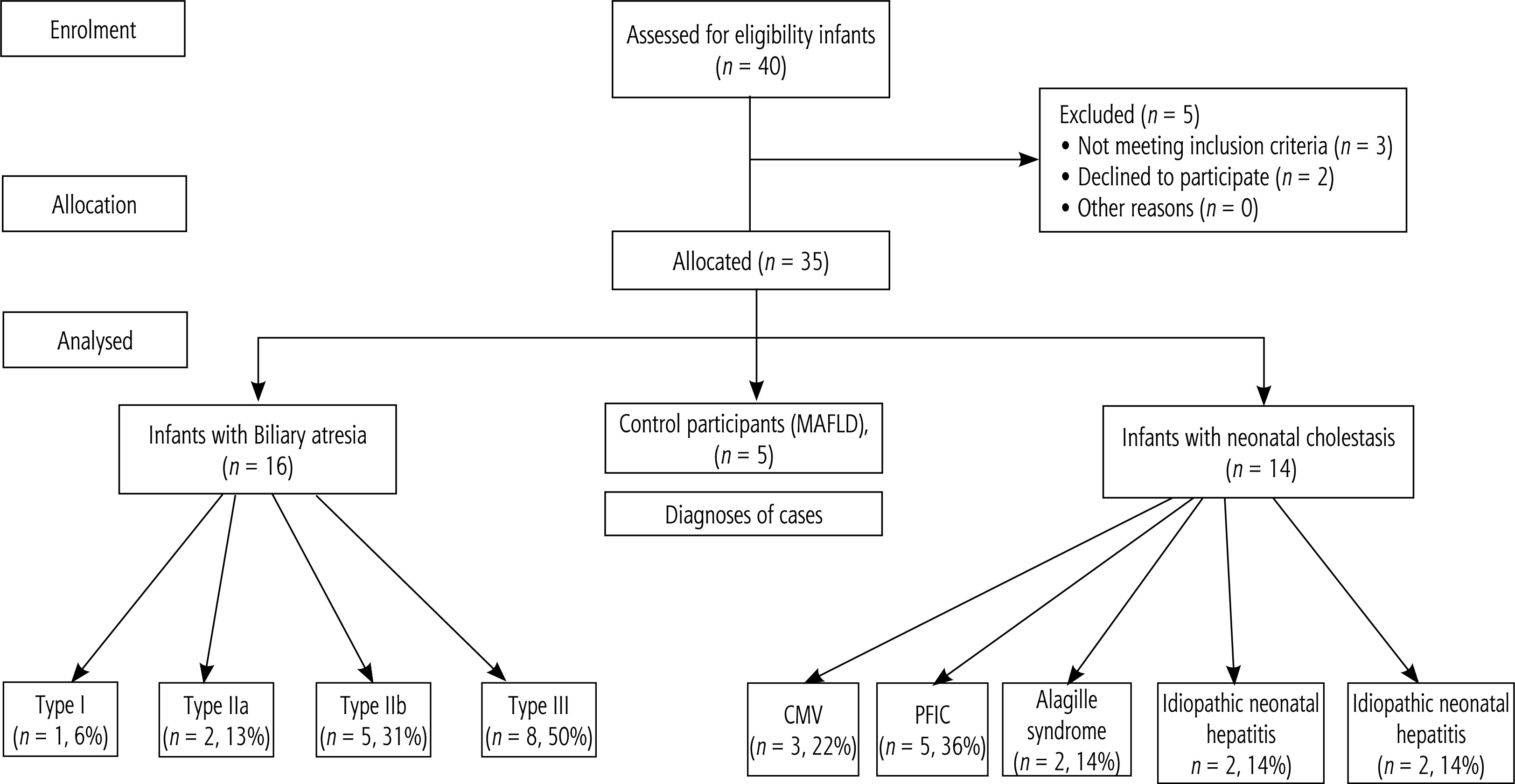
Patients in our study were divided into 3 groups as 16 infants with BA (45.7%), 14 infants with NC other than BA (40%) and 5 control cases (14.3%). A flowchart of the studied infants is shown in Figure 1. Forty infants visited the pediatric hepatology, gastroenterology, and nutrition clinics at Menoufia University. Thirty-five infants participated and underwent statistical analysis. Of these, 16 infants had BA (BA group), 14 infants had neonatal cholestasis other than BA (NC group), and the other 5 were control cases (control group). Two infants refused to participate in the study, and three infants did not meet the inclusion criteria (control group).
Demographic, clinical and laboratory investigations of study groups
Patients in our study had a median age of 2 months and a female predominance (p = 0.334). The cases had a median total bilirubin level of 12 mg/dl at initial presentation with newborn cholestasis. According to categories of the patients, 45.7% had BA, 40% had other causes of newborn cholestasis and 14.3% were controls (Table 1). Regarding BA types, one patient had type I BA (6%), two patients had type IIa BA (13%), five patients had type IIb BA (31%) and eight patients had type III BA (50%). Only 5 patients had progressive familial intrahepatic cholestasis (PFIC) (36%), 2 patients had parenteral nutrition-induced cholestasis (14%), 2 patients had Alagille syndrome (14%), 2 patients had idiopathic neonatal hepatitis (14%) and 3 patients had cytomegalovirus-induced cholestasis (22%) (Fig. 2).
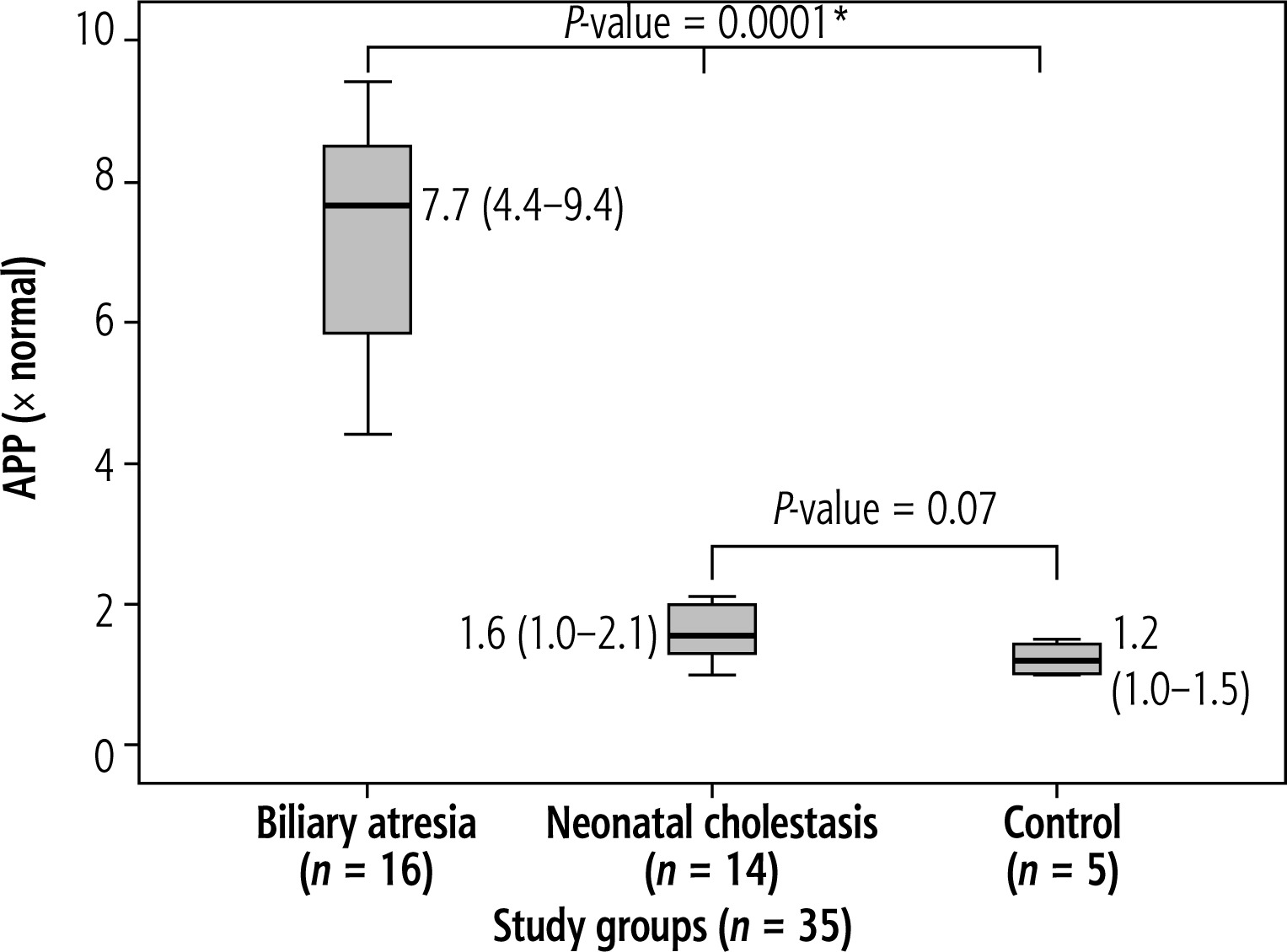
Table 1
Demographic, clinical and laboratory characteristics of study groups
| Parameter | BA (n = 16) Median (range) or n (%) | NC (n = 14) Median (range) or n (%) | P-value |
|---|---|---|---|
| Age (days) | 71 (45-105) | 63 (45-107) | 0.334 |
| Sex | |||
| Male | 6 (37.5) | 6 (42.9) | 0.529 |
| Female | 10 (62.5) | 8 (57.1) | |
| INR | 1.1 (0.9-1.3) | 1.2 (0.9-2.2) | 0.038* |
| Hb (gm/dl) | 9.4 (8.7-12.7) | 9.7 (6.2-13.2) | 0.667 |
| WBCs (× 103/dl) | 15.3 (6.9-272) | 11.9 (8.7-28) | 0.275 |
| Platelets (× 103/dl) | 473 (251-778) | 406 (157-540) | 0.154 |
| TB (mg/dl) | 9.6 (5.9-18.2) | 9.1 (5.1-15.8) | 0.484 |
| DB (mg/dl) | 6.1 (3.1-13.9) | 5.8 (3.1-9) | 0.552 |
| AST (IU/l) | 230 (98-(592 | 268 (111-585) | 0.166 |
| ALT (IU/l) | 171 (42-511) | 207 (73-786) | 0.313 |
| ALP (IU/l) | 653 (254-1163) | 487 (57-1086) | 0.154 |
| GGT (IU/l) | 813 (11-1773) | 227 (43-1016) | 0.006* |
| Total protein (g/dl) | 5.8 (4.8-8.7) | 5.8 (4.8-6.9) | 0.498 |
| Serum albumin (g/dl) | 4 (3.3-6.7) | 3.9 (2-4.4) | 0.697 |
| Liver size | |||
| Average | 0 (0.00) | 16 (100) | 0.014* |
| Hepatomegaly | 9 (64.3) | 5 (35.7) | |
| Spleen size | |||
| Average | 14 (87.5) | 12 (85.7) | 0.648 |
| Splenomegaly | 2 (12.5) | 2 (14.3) | |
Age, clinical and laboratory characteristics compared between BA and control groups using Mann-Whitney U test, sex, liver, and spleen size compared between BA and control groups using χ2 test;
Expression of amyloid β precursor protein gene in the studied groups
In the current study, comparing BA patients to other NC patients and the control group, there was a statistically significant rise in APP expression in BA patients (p = 0.0001). However, there was no statistically significant difference in the expression of APP between the NC and control groups (p = 0.07) (Fig. 3).
Effect of age and sex on amyloid β precursor protein expression
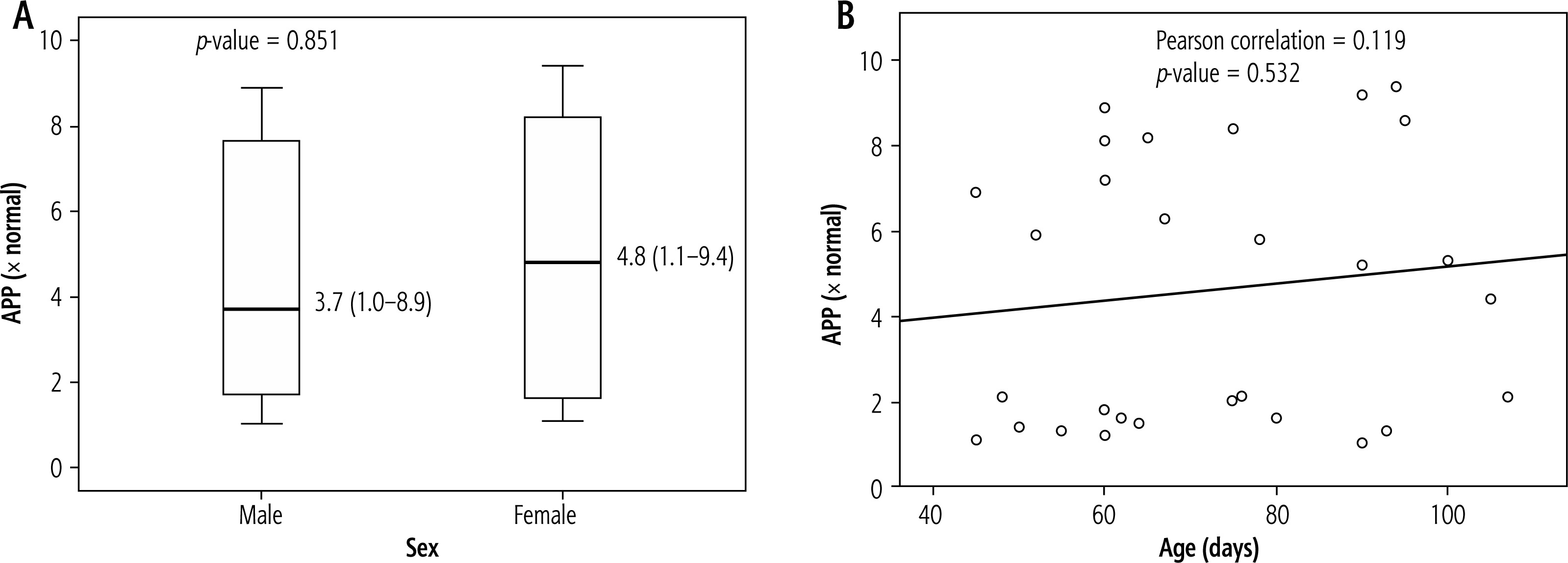
In terms of APP expression, there was no statistically significant difference between boys and girls (p = 0.851). Additionally, age and APP expression did not correlate statistically significantly (p = 0.532) (Fig. 4).
Correlation between amyloid β precursor protein expression and GGT, ALP, AST and TB
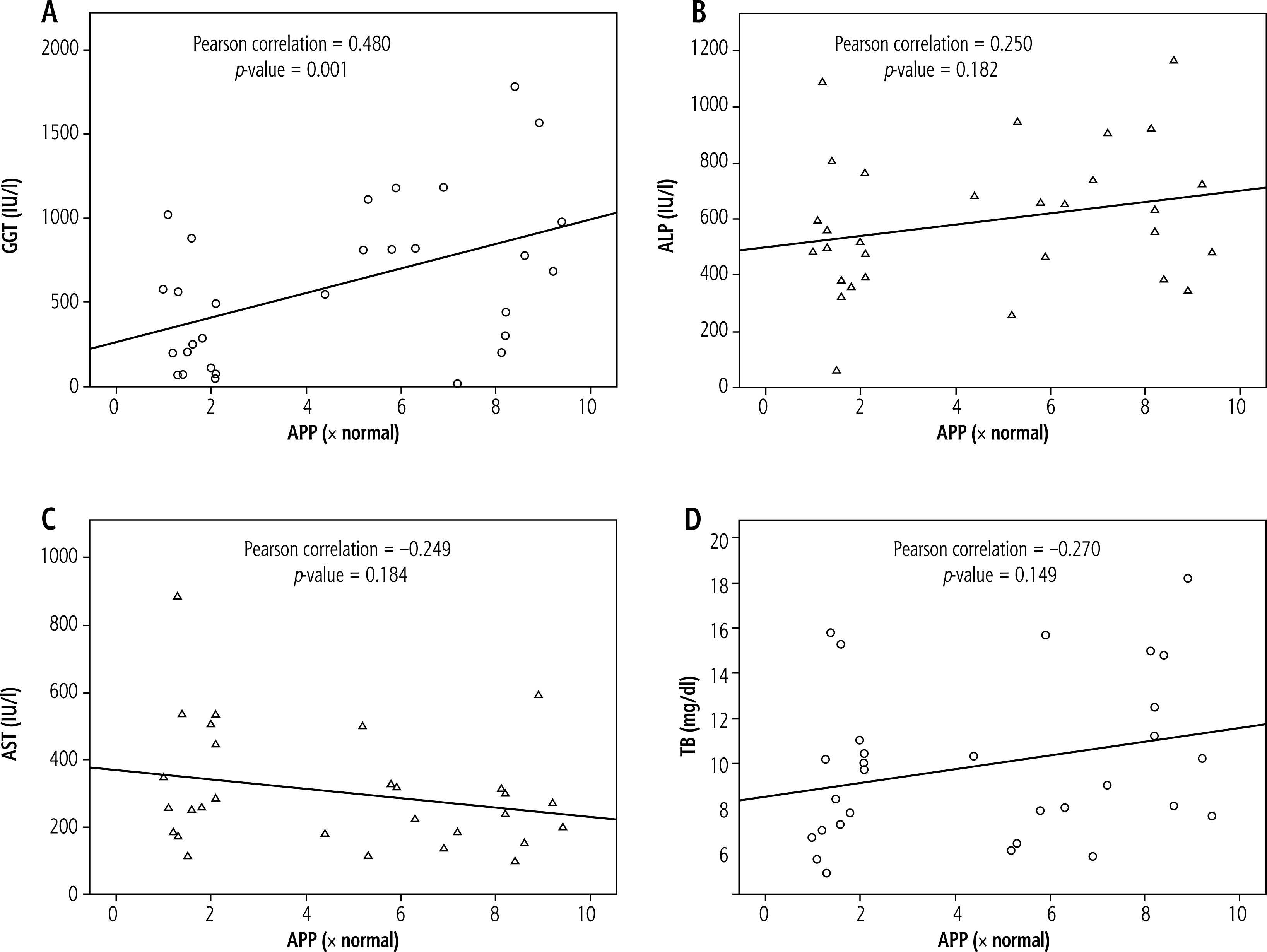
Amyloid β precursor protein showed a statistically significant positive correlation with GGT (p = 0.001). ALP, AST, and TB did not show any statistically significant correlations with APP expression (p > 0.05) (Fig. 5).
Discussion
Amyloid β precursor protein expression may influence the etiology and diagnosis of the condition in BA. Biliary atresia had greater levels of APP expression compared to other causes of NC and control groups [22]. In the present investigation, APP expression was higher in BA, pointing to both a pathogenic and diagnostic role for BA. Organoids develop an aberrant shape due to peptide accumulation in the bile ducts of BA livers. Periductal Aβ deposition and the aberrant organoid shape are the specific pathological and diagnostic characteristics of BA. Alzheimer’s disease (AD) routes seem to entail deposition. Deposition of APP was also found as the key pathogenic indication of AD [23]. What part Aβ plays in BA’s development is unclear. APP was not found in livers with other NC, except in people with BA. APP is viewed as a diagnostic tool for BA [15].
According to this study, the GGT level is higher among patients with BA compared to the NC group. In a recent study, Chen et al. [24] and Wang et al. [25] discovered that the liver enzyme GGT was elevated in patients with biliary obstruction, which can aid in diagnosis of BA, albeit not entirely.
In the current study, APP gene expression was more prevalent in BA patients than NC patients or controls. There was a significant increase in APP expression in BA patients compared to other NC patients and the control group. In the NC and controls, no statistically significant difference in the expression of APP was found. Because Aβ deposits were exclusively observed in the liver biopsies of BA patients and not in those of patients with choledochal cysts (CC), cholestasis (CS), or hepatoblastoma (HB), Babu et al. [26] concluded that they might be employed as a potential diagnostic marker for BA. Moreover, they found that the bile ducts of BA patients exhibited Aβ accumulation. The association between Aβ buildup and BA was supported by transcriptomic analysis of liver biopsies and liver organoids of BA patients, which reveals altered expression of numerous genes involved in APP processing. Similar to liver organoids from BA patients, human control organoids could also have the same problems after being exposed to exogenous Aβ. The latter finding may indicate that bile duct illness in BA is brought on by Aβ accumulation. Moreover, Tian et al. [18] discovered that in neonates with BA, Aβ expression was elevated in the blood and livers. By reducing mitochondrial respiration and changing mTOR signaling, Aβ reduced mitochondrial respiration in liver organoids. Aβ is often considered to be a useless catabolic waste. It serves a physiological antibacterial function in the body when used in smaller doses [27]. Identified Aβ deposition represents the main pathological feature of Alzheimer’s disease and cerebral amyloid angiopathy [28], around BA bile ducts [26]. This finding reveals a novel pathomechanism for BA important for future diagnosis and treatment considerations.
In the current experiment, a statistically significant correlation was found between GGT and the production of the Aβ precursor protein. ALP, AST, or TB expression did not significantly correlate with Aβ precursor protein levels. In other hand, Lyu et al. [23] found that Aβ42/Aβ40 showed sensitivity and specificity for the diagnosis of BA when combined with GGT or TB.
Detection of cholestatic liver illness is aided using hepatic function markers, such as ALT, AST, and GGT and various forms of bilirubin (TB and indirect bilirubin [IBIL]). It is consistent with the use of these hepatic function indicators in the diagnosis of cholestasis that plasma values of ALT, AST, GGT, TB, DB, and IBIL have predictive value for cholestasis. Using Pearson correlation analysis, it was discovered that A42/A40 had favorable relationships with GGT and TB [23].
Cholestasis can be predicted by the positive association between Aβ42/Aβ40 and indicators of liver function. More importantly, combinations of Aβ42/A40 and indicators of liver function significantly increased the diagnostic accuracy of Aβ42/A40 and all liver function parameters for cholestasis, suggesting that Aβ42/Aβ40 may be an additional biomarker that improves the accuracy of indicators of hepatic function common in diagnosis of biliary liver disease [23].
In this study, no significant relation of fibrosis with APP expression was found. The mechanisms in the liver are less well understood, despite the fact that the synthesis and destruction of Aβ in the central nervous system (CNS) have been widely reported [29]. Despite the knowledge that the liver generates, absorbs, and eliminates Aβ peptides, little is known about its role in both healthy and cirrhotic livers [30]. Recent research by Wiest et al. [31] discovered that Aβ42 levels in patients with liver cirrhosis are unrelated to measures of liver function. Another study by Wang et al. [32] found higher levels of Aβ42 in those with liver cirrhosis compared to healthy controls. This does in fact imply that the healthy liver controls Aβ42 levels.
It is crucial to identify new non-invasive biomarkers to enable early baby screening for BA vs. non-BA. However, our data indicated that APP was a marker for liver disease. Similar to this result, there was insignificant difference between BA and non-BA using the hepatic function measures individually or collectively [17]. However, APP increased the accuracy of BA prediction when compared to the use of other liver indices, regarding the elevated liver enzyme GGT in patients with biliary tract obstruction, and plays a significant but not solely determining role in the diagnosis of BA [33]. The addition of one of the other liver function tests to the combination of APP and GGT may further enhance the capacity to distinguish the source of cholestasis as BA or non-BA [23]. Considering everything mentioned above, our early results suggested that APP expression might be a useful adjuvant biomarker for BA diagnosis.
Limitations of the study
Larger sample sizes in subsequent research are required to support our findings because the current study’s lower sample size is insufficient. Our findings on the expression of APP are positive and corroborate those from earlier studies conducted on tissue and organoids. However, further evaluation and validation in a separate larger patient cohort of BA, non-BA cholestatic liver diseases, and conditions unrelated to the liver in multiple centers are required before the diagnostic efficacy and usefulness of APP as an adjuvant invasive biomarker in conjunction with other liver function parameters for BA, non-BA cholestatic liver diseases, and conditions unrelated to the liver are implemented into routine clinical practice.
Conclusions
We concluded that the development and identification of BA may depend on the liver expression of serum APP. Surgeons may be able to perform early intraoperative cholangiography for BA confirmation if the combination of APP with GGT and other hepatic function parameters exhibits a high predictive potential as a diagnostic test for BA. To evaluate this hypothesis, more research with sizable sample numbers is necessary.