






Introduction
Urinary bladder cancer is the most common malignancy of the bladder, and more the 90% of all bladder cancer cases are urothelial carcinoma [1]. The majority of the cases are non-invasive urothelial carcinoma [1], and only 25% of them are detected as muscle-invasive carcinoma with a stage greater than pT2 [1–3]. Most urothelial carcinoma are located directly within the urinary bladder and only the minority of 5–10% develop within the upper urinary tract [4]. In 2022, bladder cancer was the ninth most common tumour entity worldwide [5], and higher disease rates were observed in western industrial countries [5, 6]. Incidence and mortality are 2- to 6-fold increased in men compared to women [3, 7]. The most substantial risk factors for developing bladder cancer are tobacco smoking and exposure to chemical substances like aromatic amines and polycyclic aromatic hydrocarbons [6, 8, 9]. For other risk factors like diets, exposure to diesel exhaust, or an imbalanced microbiome, only correlative evidence exists [6, 9]. Therapeutic options for the treatment of bladder cancer include transurethral resection of a bladder tumour, cystectomy, and adjuvant and neoadjuvant chemotherapy, among others [10].
Keratins are important components of intermediate filaments in epithelial cells [11–14]. Intermediate filaments are, among microtubules and microfilaments, one of the main components of the cytoskeleton of the cell. They have a typical size of about 10 nm and show a specific expression pattern that depends upon the cell type, the tissue, and the differentiation status [11, 14, 15]. Importantly, during tumourigenesis, the keratin expression pattern of the original cell remains usually largely unchanged, a fact that can be used by pathologists when assessing tumour samples [11, 15]. Indeed, since their discovery in the 1950s keratins are part of the immunohistochemical panels routinely used in pathological diagnosis [14]. Furthermore, numerous studies have shown that the aberrant expression of certain keratins has prognostic value in different tumour entities [11, 14, 16, 17]. For example, the expression of CK14 in muscle-invasive urothelial carcinoma is significantly increased and the expression of CK17 significant decreased compared to the non-invasive papillary urothelial carcinoma [18]. In line with this, high expression of CK14 was significantly correlated with a shorter disease-specific survival [18]. In addition, in vitro experiments revealed a role for the keratins CK5, CK14, CK20, and CK13 for the development of resistance against the chemotherapeutic agent cisplatin in urothelial carcinoma cell lines [19].
In the present study, we analyse the expression of the 3 keratins CK10, CK13, and CK14 in 4 urinary bladder transitional cell carcinoma cell lines via western blot. Using siRNAs directed against CK13 and CK14, we downregulate protein expression of the 2 keratins in different cell lines. Furthermore, we analyse cell viability of the tumour cell lines to determine whether the downregulation of certain keratins affects tumour cell viability.
Material and methods
Reagents
The following primary antibodies were used: β-actin (A5541) from Sigma-Aldrich (St. Louis, MO, USA). CK10 (ab76318), CK13 (ab92551), and CK14 (ab181595) all from Abcam (Cambridge, United Kingdom). Secondary antibodies were anti-mouse IgG, HRP-linked antibody, anti-rabbit IgG, HRP-linked antibody (both from Cell Signaling Technology, Danvers, MA, United States) and goat anti-rabbit IgG (H+L), HRP (from Thermo Fisher Scientific, Waltham, MA, United States). The following siRNAs were used: SASI_Hs01_00066819 and SASI_Hs01_00220782 (both targeting KRT13); SASI_Hs01_00198716 and SASI_Hs01_00198719 (both targeting KRT14); as negative control MISSION® siRNA Universal Negative Control #1 (all from Sigma-Aldrich).
Cell lines and cell culture
The cell line Cal-29 was from DSMZ (Leibniz Institute DSMZ – German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany). The cell line J82 was a kind gift from Prof. Arndt Hartmann (Institute of Pathology, University Clinic Erlangen, Germany). The cell lines RT-112 and T-24 were described previously [20]. J82, T-24, and RT-112 cells were cultured in RPMI 1640 medium, and Cal-29 cells were cultured in Dulbecco’s Modified Eagles Medium (all from PAN-Biotech). The media were supplemented with 10% foetal bovine serum (Thermo Fisher Scientific, Waltham, USA), 100 U/ml penicillin, and 100 µg/ml streptomycin (PAN-Biotech, Aidenbach, Germany). All 4 cell lines were kept at 37°C and 5% CO2 in a standard incubator with a water-saturated atmosphere.
Transfection of cell lines with siRNAs
A total of 90,000 cells were seeded in a 12-well plate in a final volume of 1 ml per well. Cells were either transfected with mock control, the negative control siRNA, one of the two targeting siRNAs, or a combination of both targeting siRNAs using 1 µM siRNA and 2 µl of MISSION® siRNA Transfection Reagent (Sigma Aldrich) 30 min after seeding. Cells were then incubated at 37°C for 24, 48, or 72 hours. Afterwards, supernatant was removed, and cells were washed once with 1 ml PBS. 50 µl lysis buffer (10 mM Tris-HCl, 140 mM NaCl, 5 mM EDTA, 1% Triton-X 100, protease inhibitor cocktail, and phosphatase inhibitor cocktail (both Sigma-Aldrich), pH 7.5) were added to the cells, and the cells were incubated on ice for 20 min. Afterwards, the remaining cells were detached, and the cell suspension was collected, placed in a reaction tube, and centrifuged. The supernatant, corresponding to the cell lysate, was transferred to a new reaction tube, and the protein amount was determined using the BIO-Rad DCTM Protein Assay (Bio-Rad Laboratories, Hercules, California, United States). Cell lysates were stored at –80°C until further use.
Western blotting
Western blotting was performed with slight modifications as described previously [21]. Usually, 35 µg protein of total cell lysates per lane were separated by SDS-PAGE on 12.5% gels and transferred to 0.2 µm nitrocellulose membranes. Successful transfer of proteins was ensured by staining of the membranes with ponceau staining. Membranes were blocked in tris-buffered saline supplemented with 5% skimmed milk powder for 1 hour at room temperature. Afterwards, the membranes were washed repeatedly with TBS-T (Tris-buffered saline supplemented with 0.25% tween-20) and incubated with the primary antibodies at 4°C overnight. Primary antibodies were diluted as follows: CK10 1 : 10,000; CK13 1 : 100,000; CK14 1 : 20,000 (all diluted in TBS-T with 5% bovine serum albumin); β-actin 1 : 10,000 (diluted in TBS-T with 5% skimmed milk powder). After washing 3 times with TBS-T, the membranes were incubated with secondary antibodies (diluted 1 : 1000 in TBS-T supplemented with 5% milk powder) for one hour at room temperature. Membranes were again washed with TBS-T and afterwards incubated with immobilon western HRP substrate (Millipore by Merck, Darmstadt, Germany) and detected using the ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories). After the addition of 20 ml Stripping Puffer (RestoreTM Western Blot Stripping Buffer, Thermo Scientific) for 10 min at room temperature to remove bound antibodies, membranes were washed 3 times for 5 min with TBS-T, blocked again, and incubated with antibodies to detect β-actin as loading control, as described above.
Cell viability assay
Cell viability was analysed using the CellTiter-blue assay (Promega, Fitchburg, WI, USA) as described previously [22, 23]. In brief, 90,000 cells were seeded in duplicates in a 12-well plate, transfected with siRNAs or mock, and incubated at 37°C for 72 hours. Afterwards, the reaction reagent was added, and the fluorescence intensity was measured at 590 nm using FLUOstar Omega (BMG Labtech, Ortenberg, Germany) and normalised to the value received at the starting point.
Statistical analysis
Data were analysed by one-way ANOVA following Dunnet’s multiple comparison test. Data analyses were performed using GraphPad Prism version 10 (GraphPad Software, La Jolla California USA, www.graphpad.com). We performed a comparison of cells transfected with the negative control siRNA with all other conditions. Statistical significance was determined at a level of p < 0.05. Further details are also given in the respective figure legends.
Results
Expression of CK13, CK14, and CK10 in different human urinary bladder carcinoma cell lines
Keratins show cell-type-specific expression patterns. Importantly, aberrant expression of keratins has been reported in several different tumour entities including cancers of the urinary bladder, and expression profiles of keratins therefore can have diagnostic or even therapeutic value. For this study, we used the 4 urinary bladder transitional cell carcinoma cell lines Cal-29, J82, T-24, and RT-112.
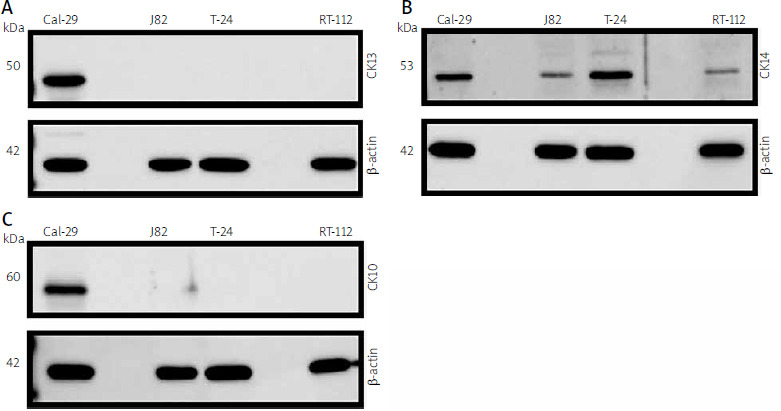
Cal-29 was established from a muscle-invasive, simultaneously osseus metastatic urothelial carcinoma (G4, pT2) [24], and J82 was established from a poorly differentiated, muscle-invasive urothelial carcinoma (G3, pT3) [25]. T-24 was established from a high-grade urothelial carcinoma [26], whereas RT-112 was derived from tumour cells of a non-invasive urothelial carcinoma (G2, pTa) [27]. To investigate whether these cells express different keratins, we first prepared cell lysates of all 4 cell lines and analysed CK13 expression by western blot. We could detect CK13 expression in Cal-29 cells, but not in J82, T-24, or RT-112 cells (Fig. 1 A). We then analysed the same lysates for CK14 expression. In contrast to CK13, CK14 was expressed in all 4 cell lines, albeit with different protein amounts – Cal-29 and T-24 cells showed modest CK14 expression (Fig. 1 B). In contrast, CK14 expression in J82 and RT-112 cells was much weaker, but still detectable (Fig. 1 B). Afterwards, we analysed expression of CK10. Interestingly, CK10 was only detectable in the lysate of Cal-29 cells, but not in the other 3 cell lines (Fig. 1 C). In summary, our data show that CK14 is expressed in all 4 urinary bladder carcinoma cell lines, whereas CK10 and CK13 are only expressed in Cal-29 cells.
Fig. 1
Expression of CK13, CK14, and CK10 in different human urinary bladder carcinoma cell lines. Expression of CK13 (A), CK14 (B), and CK10 (C) was analysed by western blot in lysates from Cal-29, J82, T-24, and RT-112 cells. Proteins were detected by keratin-specific antibodies. Equal loading of protein amounts was ensured by re-staining the membrane after stripping with an antibody directed against β-actin. In each panel one western blot from 3 independent experiments with similar outcomes is shown
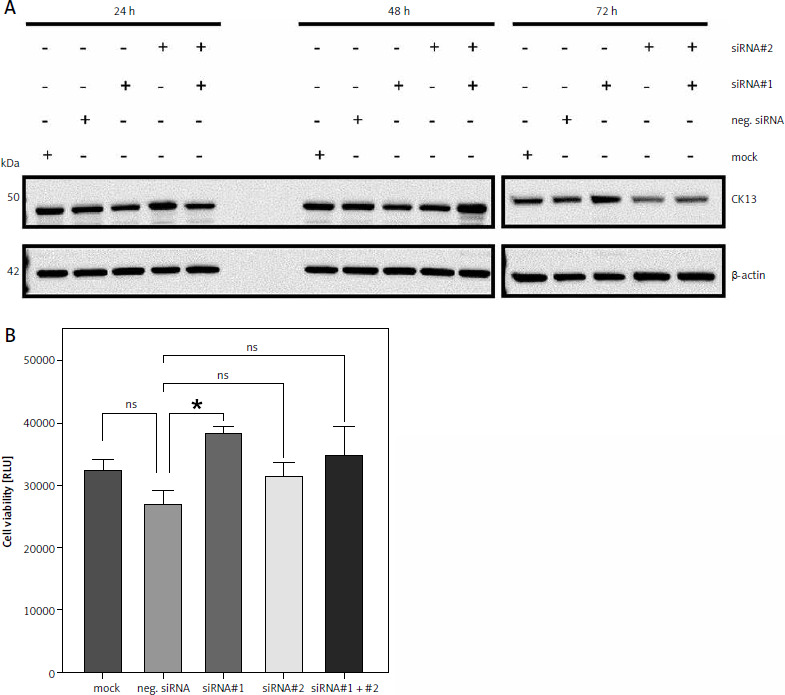
Downregulation of CK13 in Cal-29 cells does not affect cell viability
Having shown that Cal-29 cells express CK13, we wanted to investigate whether downregulation of CK13 would affect cell viability. For this, we used 2 different siRNAs targeting KRT13, the gene that encodes the CK13 protein. We transfected Cal-29 cells either with one of the two siRNAs alone, both siRNAs combined, a negative control siRNA, or a mock control without any siRNA. Because proteins have different half-lives, we tried to determine the optimal period between siRNA transfection and functional analysis of the cells and therefore lysed cells 24, 48, and 72 hours post transfection. As shown in Figure 2 A, we did not detect any significant change in CK13 abundance via western blot after 24 and 48 hours, irrespective of which siRNA was transfected. However, we noted a decrease in CK13 expression 72 hours after transfection when siRNA#2 or the combination of siRNA#1 and siRNA#2 was transfected (Fig. 2 A). Based on this, we chose to investigate cell viability of Cal-29 cells 72 hours after transfection, expecting that a functional consequence of CK13 downregulation would be apparent at this time point, but probably not after 24 or 48 hours. However, we could not detect a meaningful difference between the downregulation of CK13 and the cell viability of the Cal-29 cells, because neither the siRNA#2-transfected cells nor the cells transfected with siRNA#1 and siRNA#2 showed a significant difference in terms of cell viability (Fig. 2 B). We concluded from this that CK13 is not causatively involved in the viability of Cal-29 cells.
Fig. 2
Downregulation of CK13 in Cal-29 cells does not affect cell viability. Cal-29 cells were seeded in 12-well plates and transfected with CK13-specific siRNA#1, siRNA#2, a combination of both, a negative control siRNA, or received mock treatment without any siRNA. Cells were lysed after 24, 48, or 72 hours, and the amount of CK13 in the cell lysate was analysed by western blot. β-actin was analysed to verify equal loading. One representative experiment of 3 performed with similar outcomes is shown (A), cells were transfected as described in the legend of panel A, and a cell viability assay was performed 72 hours after transfection. Statistical analysis was performed using one-way ANOVA following Dunnett’s multiple comparison test (B). Here, all conditions were compared to the cells transfected with the negative control siRNA
* p < 0.05
ns – no significant difference
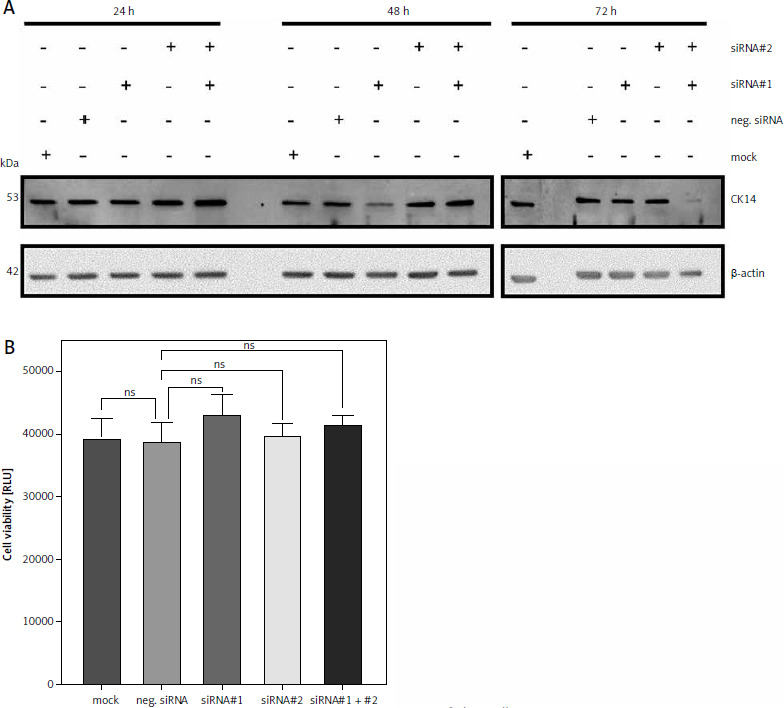
Downregulation of CK14 does not affect cell viability of Cal-29 cells
Having shown that CK13 is not involved in cell viability of Cal-29 cells, we sought to investigate whether this might be the case for CK14. We had previously established that CK14 is expressed in Cal-29 cells (Fig. 1 B) and therefore now employed a similar strategy as described above to investigate its role in cell viability. For this, we again transfected Cal-29 cells with either 2 different siRNAs targeting KRT14, the gene encoding CK14, a combination of both siRNAs, a negative control, and a mock control lacking a siRNA. Similarly, we analysed CK14 expression 24, 48, and 72 hours after transfection of the siRNAs via western blot. Like for CK13, we could not detect any influence of the siRNAs on the amount of CK14 24 hours after transfection (Fig. 3 A). Although we noted a small decrease of CK14 protein 48 hours after transfection of siRNA#2, the strongest reduction in CK14 protein levels was again achieved after 72 hours and the transfection of the 2 different siRNAs together (Fig. 3 A). Therefore, we conducted the experiment regarding the cell viability also 72 hours after transfection. However, like for the downregulation of CK14, we did not detect a significant difference between the negative siRNA control compared to any of the siRNA-transfected cells, arguing against a functional role of CK14 in cell viability (Fig. 3 B).
Fig. 3
Downregulation of CK14 does not affect cell viability of Cal-29 cells. Cal-29 cells were seeded in 12-well plates and transfected with CK14-specific siRNA#1, siRNA#2, a combination of both, a negative control siRNA, or received mock treatment without any siRNA. Cells were lysed after 24, 48, or 72 hours and the amount of CK14 in the cell lysate was analysed by western blot. β-actin was analysed to verify equal loading. One representative experiment of 3 performed with similar outcomes is shown (A), cells were transfected as described in the legend of panel A, and a cell viability assay was performed 72 hours after transfection. Statistical analysis was performed using one-way ANOVA following Dunnett’s multiple comparison test. Here, all conditions were compared to the cells transfected with the negative control siRNA. ns: no significant difference (B)
ns – no significant difference
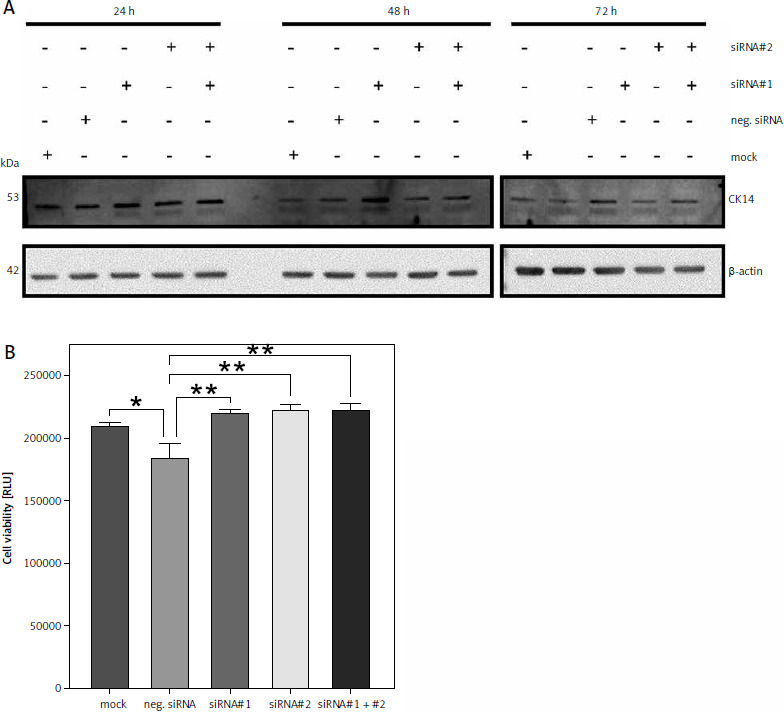
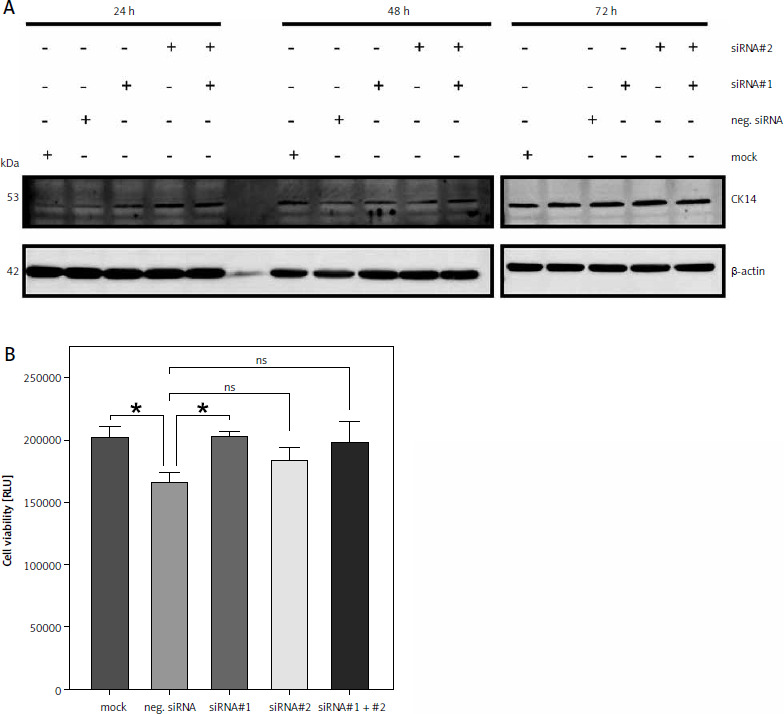
Downregulation of CK14 does not affect cell viability of T-24 cells
Because neither CK13 nor CK14 appear to be involved in cell viability of Cal-29 cells, we sought to verify this observation in a second urinary bladder carcinoma cell line. CK14 is the most abundant keratin that we have investigated in our initial analysis, and it is highly expressed in T-24 cells (Fig. 1 B). Therefore, we repeated the experiments described above with T-24 cells. As shown in Figure 4 A, downregulation of CK14 was rather unsuccessful, as we could not observe a downregulation of CK14 after 24 or 48 hours, irrespective of whether we transfected the cells with the single siRNAs or a combination of both. And although the CK14 amounts appear to be reduced 72 hours after transfection, this was also true for cells treated with the mock control or transfected with the negative siRNA (Fig. 4 A). Nevertheless, we performed a cell viability assay 72 hours after transfection of the cells. However, we could not detect a reduction in cell viability when siRNAs targeting KRT14 were transfected. Rather, the negative control showed a small but significant reduction in cell viability compared to all other conditions, including the mock control (Fig. 4 B). We concluded from these results that CK14 plays also no major role in the viability of T-24 cells.
Fig. 4
Downregulation of CK14 does not affect cell viability of T-24 cells. T-24 cells were seeded in 12-well plates and transfected with CK14-specific siRNA#1, siRNA#2, a combination of both, a negative control siRNA, or received mock treatment without any siRNA. Cells were lysed after 24, 48, or 72 hours and the amount of CK14 in the cell lysate was analysed by western blot. β-actin was analysed to verify equal loading. One representative experiment of 3 performed with similar outcomes is shown (A), cells were transfected as described in the legend of panel A, and a cell viability assay was performed 72 hours after transfection. Statistical analysis was performed using one-way ANOVA following Dunnett’s multiple comparison test (B). Here, all conditions were compared to the cells transfected with the negative control siRNA
* p < 0.05
** p < 0.01
ns – no significant difference
Unsuccessful downregulation of CK14 in J82 cells
Our initial screening showed that CK14 is expressed in J82 cells, but to a much smaller amount compared to T-24 and Cal-29 cells (Fig. 1 B). Because our results so far revealed no functional role for CK14 for cell viability and we were not able to completely downregulate CK14 expression, we wondered whether we would be able to achieve better results when we used a cell line with less CK14 expression. Therefore, we performed a similar experiment with J82 cells and tried to downregulate CK14 using the same 2 siRNAs we had used in previous experiments. However, we could not detect any significant influence of the siRNAs on the CK14 amount in the lysate of J82 cells, irrespective of whether we harvested the cells 24, 48, or 72 hours after transfection (Fig. 5 A). For the sake of completeness, we also performed a cell viability assay 72 hours after transfection, but again we could not detect any meaningful differences between the 5 conditions tested (Fig. 5 B). In summary, although J82 cells express lower amounts of CK14 protein compared to the other cell lines investigated in this study, we have not been able to successfully downregulate this amount via siRNA-mediated targeting.
Fig. 5
Unsuccessful downregulation of CK14 in J82 cells. J82 cells were seeded in 12-well plates and transfected with CK14-specific siRNA#1, siRNA#2, a combination of both, a negative control siRNA, or received mock treatment without any siRNA. Cells were lysed after 24, 48, or 72 hours and the amount of CK14 in the cell lysate was analysed by western blot. β-actin was analysed to verify equal loading. One representative experiment of 3 performed with similar outcomes is shown (A, cells were transfected as described in the legend of panel A, and a cell viability assay was performed 72 hours after transfection. Statistical analysis was performed using one-way ANOVA following Dunnett’s multiple comparison test (B). Here, all conditions were compared to the cells transfected with the negative control siRNA
* p < 0.05
ns – no significant difference
Discussion
Bladder cancer is among the 10 most common tumour entities worldwide [5]. A curative treatment for muscle-invasive bladder cancer is still difficult, and new therapeutic options have yet to be developed. Furthermore, diagnostic markers that can be used to identify tumour tissue, differentiate between different tumour subtypes, or have prognostic value in terms of disease-free or overall survival are needed. The expression profiles of different keratins are used for decades in several tumour entities, including bladder cancer [28]. However, several keratins have not been evaluated in this regard and therefore represent potential novel diagnostic markers or even therapeutic targets.
In the present manuscript, we analysed 4 different human urinary bladder carcinoma cell lines. We showed that CK14 is expressed in all cell lines, whereas CK10 and CK13 are only detectable in the Cal-29 cell line and are absent in the 3 other investigated cell lines. Cal-29 was established in 2001 from a muscle-invasive urothelial carcinoma (G4, pT2) [24] and thus represents a high-grade tumour in our in vitro assays. This is of course suggestive that high-grade tumours of the bladder change their keratin expression profile by upregulation of CK10 and CK13, but this might also be a unique characteristic of this cell line. Although we included 4 different cell lines representing different stages of bladder cancer, one must be aware that cell lines are never able to fully represent the diversity of primary human tumour samples. Thus, all conclusions drawn from the results of this study must be confirmed and replicated in future studies using immunohistochemical stainings of tumour samples from human patients.
We further investigated whether downregulation of CK13 or CK14 affects cell viability of the tumour cell lines. We used different siRNAs targeting either CK13 or CK14, and although we were able to reduce the protein amounts in most cell lines, the reduction was rather modest and never complete. In J82 cells, we were not able to reduce CK14 levels at all. Because proteins have different turnovers and half-lives within the cell, we determined the efficiency of our knockdown after 24, 48, and 72 hours, and we could only see results after 72 hours. In contrast, a recent study showed knockdown of KRT17 by siRNA in gastric cancer cells, which induced antitumoural effects like significantly reduced cell proliferation, the inhibition of cell migration and invasion, and a reduction of tumour growth in vivo [29]. In contrast to our approach, the authors transfected siRNAs 24 hours after seeding the cells, which might be a better option also for urinary bladder carcinoma cell lines and should be evaluated in the future. Interestingly, downregulation of KRT17 was achieved in T24, J82, and TCCSUP cells via a lentiviral-delivered shRNA, which showed most efficiency 48 hours after transduction [30]. Furthermore, knockdown of KRT13 was achieved via a shRNA in breast cancer cells [31].
Importantly, our data do not support a functional role for CK13 and CK14 in the investigated cell lines because no significant influence on cell viability could be determined in siRNA-transfected cells. This might either stem from the incomplete downregulation of the keratins, or simply imply that CK13 and CK14 are not major factors for cell viability in urinary bladder carcinoma cells. Another possibility is that the decrease in the amount of CK13 and/or CK14 needs more time than the examined 72 hours, and experiments lasting 96 hours or more might have a greater effect on the cell viability. Further evaluation of other keratins in these cell lines or the development of a better experimental strategy to downregulate CK13 and CK14 protein amounts might answer the question of whether keratins can be used as therapeutic targets in bladder cancer.
Conclusions
Our data show that human urinary bladder carcinoma cells expressed CK14, while CK13 and CK10 were only expressed in one of 4 cell lines tested. Downregulation of CK13 and CK14 does not result in impaired cell viability, suggesting that CK13 and CK14 are not valuable targets for therapeutic intervention. In future experiments CK10, CK13, and CK14 should be stained in tumour sections to investigate whether these 3 keratins are aberrantly expressed in bladder cancer and whether their expression profile can be used to predict patient survival.