






Introduction
A summary of glioblastoma
Between 20–40% of brain tumours are primary, while 60–80% are metastatic. Among adults, malignant gliomas, such as glioblastoma and astrocytoma, are the predominant types, with glioblastoma being the most aggressive and common primary brain tumour. Glioblastoma constitutes 14.5% of all central nervous system (CNS) tumours and 48.6% of malignant CNS tumours, with a median overall survival of just 15 months [1, 2].
According to the 2021 World Health Organization (WHO) guidelines, glioblastoma without an isocitrate dehydrogenase mutation is diagnosed based on genetic parameters such as telomerase reverse transcriptase promoter mutation, epidermal growth factor receptor gene amplification, and the combined gain of chromosome 7 and loss of chromosome 10, along with microvascular proliferation and necrosis. Notably, the term “glioblastoma” now specifically refers to adult cases, excluding paediatric-type neoplasms [3]. However, existing literature on early growth response 1 (EGR1) might not distinguish between adult and paediatric gliomas, potentially deviating from the current WHO definition.
The potency of cancer stem cells in glioblastoma and the tumour’s diffuse nature complicate surgical removal, contribute to treatment resistance, and lead to high recurrence rates [4]. Among key cellular components that frequently undergo dysregulation in glioblastoma are transcription factors. Dysregulation of transcription factors contributes to various processes that may enhance or inhibit the malignant characteristics of glioblastoma. For instance, oncogenic transcription factors, such as STAT3, NF-κB, C/EBPβ, GLI1, SOX2, and TEAD, drive glioblastoma progression by enhancing cell proliferation and survival. Conversely, transcription factors like p53, Capicua, NFI A and B, Myt1, Myt1l, and brachyury inhibit oncogenic signalling in glioblastoma [5].
Early growth response 1 is a key transcription factor believed to play a dual role in glioblastoma. While EGR1 is well-known for its contributions to brain functions like cognition, synaptic plasticity, stress responses, and conditions such as schizophrenia, its role in the progression of glioblastoma remains unclear and debated, warranting further investigation [6].
An introduction to early growth response 1
Early growth response 1, also known as zinc finger protein 268 (ZNF268) or nerve growth factor-induced protein A (NGFI-A), is a transcription factor within the EGR family of Cys2His2 (C2H2)-type zinc finger proteins. It influences diverse cellular processes such as cell proliferation, differentiation, migration, apoptosis, and inflammation. The gene encoding this protein is located on the q31.2 band of chromosome 5 in humans. Early growth response 1 protein consists of 543 amino acids, with a molecular weight of 57.5 kDa. It is characterised by a C-terminal activation domain, an N-terminal, an inhibitory domain, and a DNA-binding domain [6]. The DNA-binding domain comprises 3 C2H2 zinc finger domains, each approximately 23 amino acids in length. Zinc fingers 2 and 3 (amino acids 361–419) interact with amino acids 315–330 of EGR1 for nuclear localisation. Early growth response 1 then interacts with the DNA sequence 5’-GCG(T/G)GGGCG-3’ (EGR site) on gene promoters, thereby regulating target gene transcription. The activity of EGR1 can be regulated by phosphorylation on amino acids Threonine-309, Serine-350, Serine-378, Threonine-391, and Threonine-526 sites, and SUMOylation at Lysine-272. Moreover, the transcriptional co-repressors NGFI-A binding protein (NAB) 1 and 2 inhibit EGR1 transcriptional activity by binding to the repressor domain [7].
Throughout the adult brain, EGR1 exhibits extensive expression, maintaining baseline levels in the amygdala, hippocampus, striatum, and medial prefrontal cortex, governing cognition, sensitivity to reward, social behaviour, emotional response, and stress response. Its expression dynamics during long-term potentiation and long-term depression serve as significant indicators of neuronal stimulation, contributing to synaptic plasticity and processes like learning and memory [6, 8]. Additionally, EGR1 is implicated in conditions such as neuropsychiatric disorders, anxiety, and addiction [9]. Beyond neural functions, EGR1 regulates cell proliferation across various cell types, including dermal papilla cells, rhabdomyosarcoma cells, endometrial stromal cells, lung cancer cells, and prostate tumour cells. Its role in promoting neoplasms aligns with regulating proteins crucial for cell cycle and differentiation, such as cyclin D2, P19ink4d, and Fas [8].
In response to stress or injury and some cancer types, EGR1 exhibits a dual role in cell survival. While EGR1 is essential for programmed cell death in certain scenarios, it can also stimulate cell proliferation and suppress apoptosis in other contexts, showcasing its adaptable modulation of cellular responses to various stimuli [10]. This phenomenon has been observed in hepatocarcinoma, prostate cancer, rhabdomyosarcoma, and pituitary tumours [10–14]. Several studies also suggest that this dual role may apply to glioblastoma. Therefore, this article aims to review the current understanding of EGR1’s involvement in glioblastoma, drawing on data obtained from searches for relevant literature in English articles on the PubMed and Scopus databases.
Early growth response 1 and glioblastoma
Oncogenic roles of early growth response 1 in glioblastoma
Early growth response 1 can exhibit dual roles in tumourigenesis, acting as a tumour suppressor and an oncogene factor depending on the cellular context. As a downstream protein of the mitogen-activated protein kinase signalling pathway, EGR1 can promote transcription of cyclin D1 in many tumour types and maintain the mitosis of tumour cells. Meanwhile, EGR1 contributes to tumour metastasis as well, by promoting the expression of the transcription factors snail family transcriptional repressor 1 and 2 (SNAI1 and SNAI2).
The expression of EGR1 is notably common in glioblastoma, accounting for over 80% of cases in a cohort study conducted by Sakakini et al. in 2016. In another study on 207 glioma cases, including 190 glioblastomas, EGR1 protein overexpression was found in all investigated tumours but with considerable inter-tumour variation. Early growth respon- se 1 expression was declining from the central tumour area towards the tumour periphery, which had the lowest expression, consistent with previous in silico studies. In this study, EGR1 expression did not have independent prognostic potential [15]. However, Kyani et al. in 2018, by inhibiting protein disulfide isomerase in U87MG, U118MG, NU04, and A172 human glioblastoma cell lines and studying the nascent RNAs in treated cells using bromouridine-labelling and sequencing, suggested that EGR1 may be a novel marker in glioblastoma [16]. Similarly, Ma et al. in 2021 conducted an analysis of 72 clinical samples, revealing that the expression of the long non-coding RNA (lncRNA) HNF1A-AS1 is upregulated and correlated with poor patient survival in glioblastoma through the miR-22-3p/Enolase 1 (ENO1) axis, and knockdown of this lncRNA significantly suppresses malignant phenotypes. Additionally, they demonstrated that EGR1 acts as the regulator of this process by directly binding to the promoter region of HNF1A-AS1 [17]. In addition to experimental investigations, Uthamacumaran et al. proposed, employing a graph theoretic approach, that EGR1 may not be a pivotal transcription factor dictating glioblastoma behaviour. Instead, they suggested that it represents a distinctive signature capable of accurately predicting patient groups, including paediatric and adult glioblastoma, and adult glioma-derived stem cells. Interestingly, EGR1 emerged as one of the primary “interaction transcription factor markers” in adults that was associated with regulating 2 of the 4 central genes within glioblastoma regulatory networks [18].
Existing literature indicates that EGR1 potentially plays a role in angiogenesis, cell adhesion, cell survival, resistance to chemotherapy, and stemness of glioblastoma. In most studied glioblastoma cell lines, except for UP029, hypoxia induces the upregulation of EGR1. Under this condition, EGR1 directly binds to the hypoxia-inducible factor 1α promoter, resulting in the activation of the expression of vascular endothelial growth factor A and the formation of blood and lymphatic vessels in the tumour [19]. Regarding cell adhesion, in a study conducted on U251 glioblastoma cells, it was demonstrated that EGR1 positively regulates the transcription of genes encoding fibronectin, plasminogen activator inhibitor-1 (PAI-1), and transforming growth factor β1 (TGF-β1). Consequently, glioblastoma cells expressing EGR1 exhibited significantly increased cell adhesion and migration capabilities. The authors of the study suggested that EGR1 might govern cell interaction with the extracellular matrix by orchestrating the induction of fibronectin, TGF-β1, and PAI-1 in human glioblastoma cells. They also identified EGR1 binding sites within the fibronectin gene at positions –75 to –52 and –4 to +14 [20].
An important tumour-suppressive mechanism in glioblastoma is mitophagy, which is the autophagy-dependent degradation of mitochondria. Deregulated platelet-derived growth factor receptor (PDGFR) signalling is a frequent oncogenic driver in glioblastoma. One consequence of oncogenic PDGFR signalling is the increased transcription of the METTL3 RNA methyltransferase by EGR1. METTL3 methylates RNA to generate N6-methyladenosine (m6A). This modification of m6A can regulate gene expression post-transcriptionally. In glioma stem cells, one key target of METTL3 is the mRNA encoding optineurin (OPTN), which is a well-established activator of mitophagy. Consistent with OPTN having a tumour-suppressive role in glioblastoma, analysis of tumour databases revealed that glioblastoma patients with high OPTN expression have significantly longer survival than patients with lower OPTN levels. In this way, EGR1 contributes to the inhibition of mitophagy in glioblastoma [21, 22].
Temozolomide has emerged as a main treatment option for glioblastoma; however, it also constitutes a significant factor in tumour resistance and recurrence. More than 50% of glioblastoma patients treated with temozolomide do not respond to the therapy. This lack of response is attributed to the widespread exposure to temozolomide and the highly heterogeneous and mutation-prone nature of glioblastoma [23]. It has been shown that EGR1 confers temozolomide resistance by inhibiting homeobox A10 through the extracellular signal-regulated kinase (ERK)/protein kinase B/chondroitin sulphate proteoglycan 4 signalling pathway [24, 25].
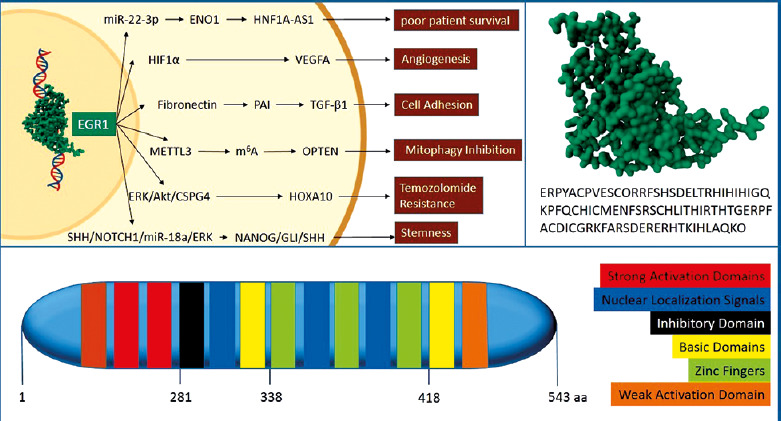
Lastly, by involvement in the signalling pathway of sonic hedgehog (SHH)/Neurogenic locus notch homologue 1 (NOTCH1)/miR-18a/ERK that is crucial for the regulation of stem cells’ self-renewal, EGR1 plays a key role in the expression and proliferation of glioblastoma stemlike cells stemness marker. Early growth response 1 regulates the expression of SHH, glioma-associated oncogene homologue 1 (GLI1), and miR-18a by direct attachment to the promoter of their genes. Early growth response 1 may also indirectly control the expression of SHH and miR-18a through a phospho-ERK-dependent mechanism, as findings on TG6 T-cell lymphoma cells demonstrate. Stimulation of these genes helps ERK to maintain phosphorylation, ultimately triggering the NANOG homeobox/GLI1/SHH regulatory network, which is essential for the clonal proliferation of glioblastoma stemlike cells [26, 27]. Figure 1 summarises the structure of the EGR1 protein and its oncogenic roles in glioblastoma.
Fig. 1
. This figure summarises the structure of the early growth response 1 (EGR1) protein and its oncogenic roles in glioblastoma. 3D structure of the zinc finger binding domain of EGR1 protein and its related amino acid sequence (A), EGR1 protein domains (B), pathways that participate in the oncogenic outcomes of EGR1 in the context of glioblastoma (C)
ENO1 – enolase 1, ERK/Akt/CSPG4 – extracellular signal-regulated kinase, protein kinase B, and chondroitin sulphate proteoglycan 4, HIF1α – hypoxia-inducible factor 1α, HNF1A-AS1 – long non-coding RNA HNF1A antisense 1, HOXA10 – homeobox A10, m6A – N6-methyladenosine, METTL3 – methyltransferase like 3, NANOG/GLI1/SHH – NANOG, glioma-associated oncogene homologue 1, and sonic hedgehog, OPTN – optineurin, PAI – plasminogen activator inhibitor, SHH/NOTCH1/miR-18a/ERK – sonic hedgehog, NOTCH1, microRNA-18a, and extracellular regulated kinase, TGF-β1 – transforming growth factor β1, VEGFA – vascular endothelial growth factor A
Early growth response 1 as a tumour suppressor
In certain cases, EGR1 acts against the development of tumours. Generally, EGR1 can increase tumour cell apoptosis by directly upregulating tumour suppressors called the non-steroidal anti-inflammatory drug-activated gene 1 and phosphatase and tensin homolog deleted on chromosome ten [26]. Other anti-tumour functions of EGR1 include invasion and metastasis inhibition by regulation of antiangiogenic proteins such as SNAI1, SNAI2, the tissue inhibitor of metalloproteinases 2, and E-cadherin [10, 28]. Due to the paucity of evidence, there is an ongoing debate about whether EGR1 acts as a suppressor of tumour growth in the context of glioblastoma [29]. One proposed idea is that the baseline expression of EGR1 might fuel tumour growth and progression. However, under certain conditions, such as when the body is under stress or when EGR1 is deliberately increased, its behaviour might switch to actively suppressing tumour growth and prompting cell death [30]. A known illustration of how cell context influences the function of EGR1 is its capacity to induce cell-intrinsic sex differences in glioblastoma. Through studies on mouse glioblastoma stem cells using bulk RNA expression analysis tools, Kfoury et al. proposed that EGR1 is among the male-biased bromodomain (Brd)-bound enhancers that operate in pluripotent stem cells specifically in males [31].
As of now, only one study has put forth a mechanism elucidating the tumour-suppressive role of EGR1 in glioblastoma. One enzyme implicated in the malignant behaviour of glioblastoma is mammalian methyltransferase-like 7B (METTL7B). METTL7B, a methyltransferase, has been identified as a factor in lowering overall survival in glioblastoma. Furthermore, depletion of METTL7B significantly reduced glioblastoma cell proliferation. Xu et al. demonstrated that knockdown of EGR1 reversed the inhibitory effect of METTL7B depletion on glioblastoma cell proliferation. Additionally, METTL7B depletion led to more glioblastoma cells arrested at the G0/G1 phase but fewer cells at the S phase, effects that were reversed by EGR1 knockdown. This suggests that EGR inhibits the contribution of METTL7B in the malignant progression of glioblastoma by an unknown mechanism [32]. This dual nature of EGR1, where it can either promote or inhibit tumour growth, depending on its expression levels and the molecular context of the cell, suggests potential therapeutic opportunities and could open avenues for targeted therapies in treating brain tumours.
Hypothetical implications of early growth response 1 in the viral hit-and-run theory
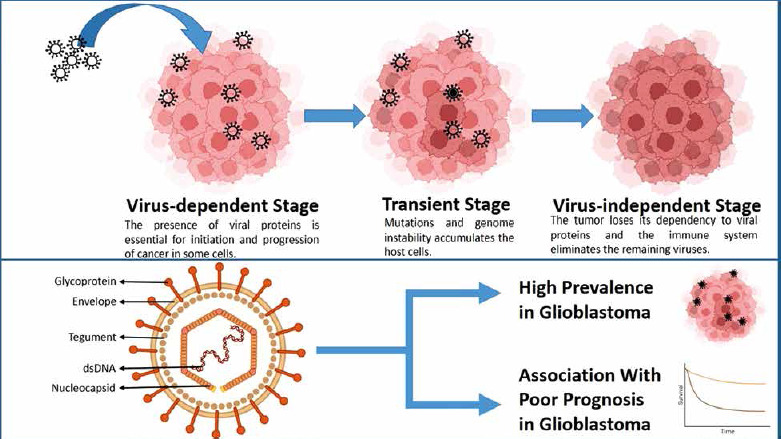
The hit-and-run theory posits that viruses aid in the accumulation of mutations and foster genomic instability until the virus is no longer necessary for tumour maintenance. Indeed, multiple studies have documented instances where tumour cells lose viral genomes, episomes, or oncogenes without losing their malignant phenotype [33]. Human cytomegalovirus (CMV) is notably implicated in causing glioblastoma through the viral hit-and-run theory [34]. One aspect of EGR1’s tumourigenic potential lies in its potential modulation of the pathology of this virus. While these implications are currently hypothetical and have not been distinctly studied, our article, aiming to offer a comprehensive review of all potential connections of EGR1 with glioblastoma, prompted us to mention these pathways as well.
Due to its high affinity for glial cells and its capacity to reduce apoptosis, increase cell invasion, activate telomerase, and enhance angiogenesis in tumour cells, numerous studies have delved into the role of CMV in glioma aetiology [35, 36]. In 2012, a group of oncologists and virologists reached a consensus, based on published data, that CMV sequences and viral gene expression occur in most if not all malignant gliomas, irrespective of WHO classification, sex, and age [37]. A meta-analysis of the findings of 32 independent studies involving over 2000 patients revealed that the detection rate of CMV in glioblastoma varied considerably due to differences between populations and detection methods. The overall estimated frequency of CMV in glioblastoma patients was found to be 62% (95% CI) [38]. Additionally, in a case-control study, researchers investigated the recursive partition analysis subclass, age, surgery, and adjuvant treatments among glioblastoma patients who survived over 18 months. Among these patients, 40% had a low CMV-positive rate, and they survived for a median of 42.5 months, suggesting a strong association between low-grade CMV infection and long-term survival in glioblastoma patients. In contrast, among patients with a shorter overall survival of less than 18 months, only 8% had a low positive rate, while 47.5% of patients had more than 75% of their tumour cells infected [39]. The genome of CMV comprises double-stranded DNA and consists of 2 large reverse domains known as long (L) and short (S). Each domain is composed of 3 regions: central unique, unique long (UL), and unique short. UL138 is one of the transcripts of the CMV genome responsible for promoting latency, regulating apoptosis, facilitating cytoplasmic assembly of viral components, and mediating TNF-α-mediated reactivation of the virus [40, 41]. Studying EGR1-mutated cells has revealed that this transcription factor plays a crucial role in the latency of CMV. Early growth response 1 binds to the CMV genome upstream of the UL138 gene and enhances the expression of this gene. Early growth response 1 is sufficient to promote UL138 expression and is currently the only known transcription factor associated with UL138 [42]. The mechanisms that occur during the latent phase of the infection are associated with several complications, including immunosenescence and malignant phenotypes, which contribute to an immunosuppressive tumour microenvironment and oncomodulation [43]. However, these observations have primarily been made in cell types other than neural cells, predominantly CD34+ haematopoietic progenitor cells. Multiple studies are required to establish such an association and its possible oncogenic role in glial cells.
Although the vast majority of research studies concerning the involvement of viruses in glioblastoma have focused on CMV, scientific attention has recently shifted to the role of Epstein-Barr virus (EBV), human herpesvirus 1 (HHV-1), HHV-2, HHV-6, human papillomaviruses, and human adenoviruses in glioblastoma pathogenesis [42, 44]. Among these viruses, EBV and HHV-1 have evidence of impactful interactions with EGR1, each with distinct mechanisms [45]. However, despite the potential of these viruses to trigger tumour activity and their reported presence in glioblastoma samples, the literature still lacks a clear association between viral infections and specific oncogenic mechanisms in glioblastoma. Figure 2 visualises the viral hit-and-run theory and the significance of CMV in glioblastoma. Additionally, Table 1 provides a concise summary of key studies investigating the roles of EGR1 in glioblastoma.
Table 1
A concise summary of key studies investigating the roles of early growth response 1 in glioblastoma, including each study’s design and main findings
| Study | Design | Findings | References |
|---|---|---|---|
| Sakakini et al. | Cohort study of glioblastoma cases | EGR1 expression was common in over 80% of glioblastoma cases | [15] |
| Kyani et al. | Experimental study on glioblastoma cell lines | EGR1 was suggested as a novel marker in glioblastoma by studying nascent RNAs in treated cells | [16] |
| Ma et al. | Analysis of 72 clinical samples | Upregulated HNF1A-AS1 correlated with poor survival; EGR1 regulates this process by binding to the HNF1A-AS1 promoter | [17] |
| Uthamacumaran et al. | Graph theoretic approach | Proposed EGR1 as a distinctive signature predicting patient groups; EGR1 regulates 2 central genes within glioblastoma networks | [18] |
| Xu et al. | Experimental study on METTL7B in glioblastoma | EGR1 knockdown reverses METTL7B depletion effects on glioblastoma cell proliferation, suggesting that EGR1 inhibits METTL7B’s malignant contribution | [32] |
| Kfoury et al. | Study on sex differences in glioblastoma stem cells | EGR1 operates as a male-biased enhancer in pluripotent stem cells | [31] |
| Multiple studies | Studies on CMV in glioblastoma | CMV sequences and gene expression in glioblastomas; CMV infection associated with shorter survival; EGR1 binds CMV genome, promotes UL138 expression, contributing to CMV latency and oncomodulation | [33–43] |
| Multiple studies | Studies on other viruses in glioblastoma | Evidence of impactful interactions with EGR1 for EBV and HHV-1 | [42, 44, 45] |
Fig. 2
In the viral hit-and-run theory, viruses aid in the accumulation of mutations and foster genomic instability until the virus is no longer necessary for tumour maintenance (A), human cytomegalovirus virion consists of a double-stranded DNA (dsDNA), nucleocapsid, tegument, a bilipid envelope layer, and surface glycoproteins (B). This virus is significantly prevalent and is linked with poor prognosis in glioblastoma cases
Conclusions
As observed in hepatocarcinoma, prostate cancer, rhabdomyosarcoma, and pituitary tumours, EGR1 appears to exhibit a dual role in glioblastoma, playing both a promoting and a suppressing role in different contexts [10–14]. Several studies suggest that EGR1 plays a significant prognostic and causative role in glioblastoma, regulating genes involved in critical processes such as cell proliferation [17], angiogenesis [19], cell adhesion [20], stemness [26], autophagy inhibition [21, 22], and temozolomide resistance [24]. However, conflicting evidence suggests that EGR1 may exert tumour-suppressive effects, as observed in studies where it suppressed the expression of METTL7B [32]. Understanding the context-dependent nature of EGR1’s actions is crucial for reconciling these contradictory findings and may have clinical implications, as observed previously in other cancers.
An example of the implications of EGR1 in cancer treatment that may apply to glioblastoma is radiation therapy. Within the promoter of EGR1, there is the distinctive CArG box sequence facilitating serum response factor (SRF) binding, a process stimulated by radiation. In prostate cancer, the activation of EGR1 during radiotherapy leads to the expression of genes like TNF-α, p53, Rb, and BAX, ultimately inducing either the arrest of tumour cell growth or cell death [46]. Given that EGR1 can trigger the activation of apoptosis-related factors and decrease the expression of survival-promoting factors, it plays a significant role in mitigating radiation resistance. In solid tumours, it is demonstrated that the level of EGR1 expression is closely associated with their response to radiation therapy. Furthermore, the expression of EGR1 in irradiated tissues shows a correlation with the remaining tumour size and the likelihood of tumour recurrence [47]. Bickenbach et al. conducted experiments wherein they fused the CArG box with the transcription start site of TNF cDNA, which incorporates the EGR1 promoter. This construct was then transfected into tumour cell lines, demonstrating its ability to significantly enhance the killing of radiotherapy-resistant tumour cells upon radiation exposure. Utilizing the CArG box within the EGR1 promoter proves to be a potent tool in anticancer strategies, particularly when combined with suicide gene therapy and radiotherapy. This approach holds immense potential for enhancing the effectiveness of therapies targeting brain tumours [48].
Based on the provided information, EGR1 shows potential as both a prognostic marker and a treatment avenue in glioblastoma. However, fully realising this potential necessitates focused research efforts to deepen our comprehension of this transcription factor and its role within the molecular framework of glioblastoma.