










Introduction
Juglone (5-hydroxy-1,4-naphthoquinone) is a phenolic compound that belongs to the class of naphthaquinones. It is a brown pigment found in nature in the leaves, shells, bark, and wood of black walnut trees (Juglans nigra L.), European walnut trees (Juglans regia L.), and other plants of the Juglandaceae family (Meskelevicius et al., 2016). Black walnut extract is used to treat acne and inflammatory diseases and as a dye for hair coloring. Juglone has been shown to cause toxicosis in ponies and walnut bark-chewing horses (Paulsen and Ljungman, 2005; Li et al., 2013). Black walnut extract has long been used in folk medicine (especially in China) to treat various types of cancers such as stomach, liver, and lung cancer (Li et al., 2013; Meskelevicius et al., 2016). Juglone shows cytotoxic properties in in vitro cell cultures (Table 1). It also exhibits antiviral, antibacterial, antifungal, and antitumor activities (Paulsen and Ljungman, 2005; Meskelevicius et al., 2016).
Table 1
Effects of juglone on cells in in vitro cultures (Paulsen and Ljungman, 2005; Meskelevicius et al., 2016)
The antitumor activity of juglone may be related to the intracellular production of reactive oxygen species (ROS) such as hydrogen peroxide, superoxide anion, hydroxyl radical, and peroxynitrite, which are usually produced in aerobic cells (Meskelevicius et al., 2016). Moderate amounts of ROS are required for normal cell physiological processes as they mediate intracellular signaling, promote cell proliferation, and are involved in cell cycle regulation. Excessive amounts of ROS induce cell death through apoptosis or necrosis, depending on the conditions of the intra- and extracellular environment (Meskelevicius et al., 2016). ROS are produced intracellularly through the effect of juglone on BCL2 (an antiapoptotic protein) and BAX (a proapoptotic protein) expression levels. Moreover, ROS induce depolarization of the outer mitochondrial membrane, which leads to the release of cytochrome c from the mitochondria into the cytoplasm and activation of the effector caspase 3 with subsequent activation of the caspase cascade (Meskelevicius et al., 2016; Ahmad and Suzuki, 2019).
The mechanism of action of juglone may also be related to its ability to inhibit the cis/trans peptidyl prolyl isomerase Pin1, which catalyzes the cis/trans isomerization of peptide bonds preceding prolyl residues (Campaner et al., 2017). This isomerase plays an important role in the stabilization and activation of the P53 protein – a suppressor of neoplastic transformation – after exposure of cells to factors that lead to DNA damage (Meskelevicius et al., 2016). It appears that this stabilization/activation is not related to the activation of the DNA damage-triggered signaling pathway, as the DNA damage-induced P53 protein phosphorylation at Ser15 was not reduced in Pin1–/– cells (lacking the PIN1 gene), while the accumulation of P53 was inhibited. Pin1-dependent accumulation and activation of the P53 protein are presumed to occur due to conformational changes induced by Pin1 in P53 binding following its phosphorylation at Ser33, Ser 46, and Ser315. Because juglone has been shown to inhibit transcription, which leads to the activation of the P53 protein, it can be assumed that juglone is responsible for the activation of the P53 protein. However, juglone also inhibits Pin1 isomerase activity, which may lead to a reduction in P53 stabilization (Ahmad and Suzuki, 2019).
Juglone also influences other cell signaling pathways, for example, it activates mitogen-activated protein (MAP) kinases, including ERK, JNK, and p38 in skin cells, glioblastoma multiforme cells, cervical cancer cells, melanoma cells, hepatocellular carcinoma cells, and smooth muscle cells (Wu et al., 2017). The influence of juglone on the modulation of the MAPK signaling pathway is explained by the activation of the apoptosis induction process (Ahmad and Suzuki, 2019). Some studies have shown that the activation of MAP kinase is dependent on the presence of ROS (Paulsen and Ljungman, 2005; Meskelevicius et al., 2016), which suggests that this dependence is due to the occurrence of the naphthaquinone redox cycle. In contrast to the activation of the MAP kinase pathways, juglone has been shown to inhibit the Akt kinase pathway (Ahmad and Suzuki, 2019). In prostate cancer cells, juglone has been shown to inhibit the Akt/GSK-3b/Snail pathway and the subsequent epithelial-mesenchymal transition. Juglone reduces the level of Akt phosphorylation resulting from high glucose levels, which leads to the suppression of the eNOS-dependent nitric oxide pathway (endothelial NOS; nitric oxide synthase 3; NOS3) in the aorta of rats (Ourique et al., 2015; Pektas et al., 2018; Ahmad and Suzuki, 2019). Juglone reduces the level of Akt phosphorylation in breast cancer cells resistant to trastuzumab (anticancer monoclonal antibody) (Sajadimajd and Yazdanparast, 2017).
The present study aimed to analyze the effect of juglone on the viability and proliferation of melanoma cells of C-32 (amelanotic melanoma) and COLO 829 (melanotic melanoma) cell lines in vitro and on the mRNA expression of the proapoptotic BAX and CASP3 and the antiapoptotic BCL2 gene.
Materials and methods
Cell lines
Human melanoma C-32 and COLO 829 cell lines and human normal dermal fibroblast HFF-1 cell line were purchased from the American Type Culture Collection (ATCC; www.atcc.org).
Cell culture condition
Cell lines were cultured according to standard mammalian tissue culture protocols and by applying a sterile technique. C-32 melanoma cells and normal dermal HFF-1 fibroblasts were cultured in DMEM medium (Lonza). COLO 829 melanoma cells were cultured in Ham’s F-12 medium. Both media were supplemented with 10% fetal bovine serum (FBS; Sigma) and 1% mixture of antibiotics (containing 10 000 U/ml penicillin and 10 000 U/ml streptomycin) (Lonza) at 37°C in humidified atmosphere of 5% CO2 in air.
Viability and proliferation assay
The effects of juglone on the proliferation and viability of C-32, COLO 829, and HFF-1 cells were estimated with crystal violet dye elution (CVDE), WST-1, and lactate dehydrogenase (LDH) assays. The CVDE assay (Crystal Violet Dye Elution Kit; Xenometrix) is used to assess the number of adherent cells, i.e., living cells in a cell culture. The amount of CV attached to the bottom of the culture vessel bound by the DNA of living cells is measured spectrophotometrically and is translated into the percentage of living cells in the cell culture. Absorbance was measured at 540 nm.
WST-1 assay (Cell Proliferation Reagent WST-1; Roche) is used to assess the metabolic activity of cells. Metabolically active, living cells show the ability to convert tetrazolium salts to formazan. This reaction requires the presence of mitochondrial dehydrogenases. The reaction is linked to the color change of the culture medium and is analyzed spectrophotometrically. The overall enzymatic activity reflects the percentage of metabolically active cells in the culture. Absorbance was measured at 440 nm.
LDH assay (LDH Cytotoxicity Detection Kit; Roche) is used to assess the number of dying cells in the culture. Dying cells release LDH into the culture medium, following the disruption of cell membrane integrity; in the conversion reaction, lactate is converted to pyruvate through the reduction of NAD+ to NADH/H+, which reduces tetrazolium salts to formazan. The effect of this reaction is observed as discoloration of the culture medium. The color intensity of the medium reflects the percentage of dying cells in the culture and can be measured spectrophotometrically. Absorbance was measured at 500 nm.
In all the above-mentioned assays, absorbance measurements were conducted using the Biogenet ASYS UVM340 instrument. All data are presented as a percentage of viable, metabolically active, and dying cells in relation to control cell cultures (cell cultures not treated with juglone). All data were calculated assuming the control as 100%. Each experiment was repeated three times. Values of P < 0.05 were considered to be significant.
The workflow for conducting the CVDE, WST-1, and LDH assays was similar. In the first step, the cells were harvested using a trypsin-EDTA solution (Gibco), and the amount of cells was determined using a Bürker chamber. Subsequently, 3000 cells were seeded onto 96-well plates (Nunc). In each well, cells were cultured in 100 μl of complete DMEM (C-32 and HFF-1 cells) or Ham’s F12 (COLO 829 cells) medium. The prepared 96-well plates were placed in a CO2 incubator for 24 h. In the second step, the medium was removed from all wells of the 96-well plates and replaced with a medium supplemented with juglone at a specified concentration (5, 25, 50, and 75 μM). Juglone was dissolved in dimethyl sulfoxide (DMSO) to make a stock solution. The concentration of DMSO was below 0.4% in all cell cultures and did not cause any detectable effect on cell growth. The lack of DMSO effect on cell cultures was also demonstrated by other authors who investigated the effect of juglone on LS-174T cells; in their study, the final concentration of DMSO in cell cultures was below 0.3% (Qiang et al., 2013). Solutions with various concentrations of juglone in the medium were prepared by a series of dilutions. Each juglone concentration was tested in triplicate. Subsequently, 96-well plates with cell cultures treated with various concentrations of juglone were placed in a CO2 incubator for 48 h. In the third step, after the incubation period, the CVDE, WST-1, and LDH assays were performed. Each test was performed according to the manufacturer’s protocols.
Preparation of cell cultures treated with juglone at 25 μM concentration
After the cells were harvested using trypsin-EDTA solution, the cell number was determined using a Bürker chamber, and 300 000 cells were then seeded onto 5 cm diameter Petri dishes. The cells of each tested cell line were seeded onto two dishes. In each dish, the cells were cultured in 5 ml of complete DMEM or Ham’s F12 medium. Next, the Petri dishes were incubated in a CO2 incubator for 24 h. Subsequently, the medium was removed from the dishes and replaced with complete DMEM or Ham’s F12 medium with the addition of juglone at 25 μM concentration or complete DMEM or Ham’s F12 medium without the addition of juglone in control cultures. All cell cultures were then incubated in a CO2 incubator for 48 h. After the incubation period, the cell culture dishes were transferred from the incubator to an inverted microscope table, and the confluence of each culture was visually assessed. The observations were recorded by acquiring microscopy images (Fig. 1).
Fig. 1
Comparison of confluence between cell cultures treated with juglone at 25 μM concentration and control cultures not treated with juglone. Cell cultures treated with juglone showed lesser confluence than control cell cultures. This effect was more evident in C-32 melanoma cell culture. Additionally, in C-32 cell culture, juglone treatment was linked with the change in cell morphology from fibroblastic to spherical. The lowest effect of juglone on cell confluence was observed in HFF-1 fibroblasts. Microscopic images [100x] were acquired after 48 h of incubation of cell cultures. Microscopic images were captured using the Nikon Eclipse TS100 microscope and NIS Elements BR 3.0 software. Scale bar = 100 mm

Next, after removing the medium from cell cultures, the cells were harvested by a cell scraper and transferred to a 2.2 ml Eppendorf tube. Subsequently, total RNA was extracted from the cells.
Total RNA extraction
Total RNA was isolated from cells by using the Quick-RNA MiniPrep kit (Zymo Research) in accordance with the manufacturer’s protocol. The quantitative and qualitative assessments of the extracts were conducted through absorbance measurements at 260 nm wavelength (assuming that 1.0 OD260 corresponds to 40 μg RNA in 1 ml of extract). Spectrophotometric measurements were performed using an HP8452A spectrophotometer (Hewlett Packard®).
Evaluation of transcriptional activity of the BAX, BCL2, and CASP3 genes using RT-qPCR
The evaluation of transcriptional activity of the BAX, BCL2, and CASP3 was performed for all individual cell cultures treated with juglone at 25 μM concentration. Reference controls were cell cultures of each cell type not treated with juglone.
To evaluate the number of mRNA copies of the BAX, BCL2, and CASP3 in the extracts of total RNA, the RT-qPCR method was used. The 18S rRNA gene was used as a constitutive reference gene. The DNA Engine OPTICON™ fluorescence detector (MJ Research) with the QuantiTect SYBR Green RT-PCR Kit (Qiagen) and specific complementary primers to amplify the sequence were used to run the reaction. Primer sequences were designed using Primer ExpressTM Version 1.0 computer software (ABI Prism) based on the data from MEDLINE (http://www.ncbi.nlm.nih.gov) (Table 2).
Table 2
Nucleotide sequences of primers used in the RT-PCR reaction
A reaction mixture for each RNA extract with a special primer set was prepared in triplicate (each data point is the mean (average) of triplicate). The RT-qPCR was performed in a 10 μl reaction mixture with 200 ng of total RNA and 0.5 μM of final primer concentration for each forward and reverse primer. The RT-qPCR protocol was as follows: 30 min RT reaction at 50°C, a 15 min PCR activation at 95°C, followed by 45 cycles of 15 s denaturation at 94°C, 30 s annealing at 60°C, and 10 s extension at 72°C. Finally, the melting curve analysis was performed to confirm RT-qPCR specificity. Negative controls with no total RNA were included in each run of RT-qPCR. The standard curve used in the evaluation was prepared based on simultaneously amplified 18S rRNA fragments in concentrations of 100, 200, 500, 1000, and 4000 copies. 18S rRNA standards were purchased from Applied Biosystems (TaqMan® DNA Template Reagents Kit). The standard curve was used to estimate the initial number of copies (copy number) of RNA template used in the RT-qPCR reaction. The results are presented as the mRNA copy number of a specific gene transcript, converted to 1 μg of total RNA in the sample.
Identification of BAX, BCL2, and caspase 3 mRNA in HFF-1, C-32, and COLO 829 cells in in vitro cultures
In all cell cultures, the specific amplification of the expected products (amplicons of BAX, BCL2, and caspase 3 mRNA transcripts) was determined by a peak in the melting curve analysis in accordance with the primer design.
Statistical analysis
Data distribution for each trial met the requirements of normal distribution (Shapiro-Wilk test) and the homogeneity of variance (Levene test and Brown-Forsythe test). For viability and proliferation assay, statistically significant differences between the analyzed mean values were determined by performing F test in analysis of variance. The control group was compared with four groups tested by Dunnett’s test. The data were presented as charts with the arithmetic mean for each sample and the corresponding standard deviations. For the data obtained through the RT-qPCR method, the control group and one of the test groups were compared using t-test, provided that the variances of these trials were homogeneous, or using t-test with separate estimation of variance (Cochran-Cox test) if they were not homogeneous (i.e., if they failed to be homogeneous). The obtained data are presented in graphs as arithmetic mean and the corresponding standard deviations. All tests were performed using Statistica version 12.0 software with the significance level of α = 0.05.
Results
Microscopic evaluation of the effect of juglone on C-32 and COLO 829 melanoma cells and HFF-1 fibroblasts in in vitro cultures
Microscopic images of cell cultures showed differences in the confluence of cell cultures after 48 h of incubation between control cultures (without juglone addition) and cultures treated with juglone at 25 μM concentration (tested cultures) (Fig. 1).
All cell cultures treated with 25 μM juglone for 48 h showed a lower confluence than control cultures not treated with juglone. Both C-32 and COLO 829 melanoma cells were more sensitive to juglone than HFF-1 fibroblasts (lower cell density in melanoma cultures treated with juglone compared to control cultures in relation to HFF-1 cultures). Additionally, the change in cell morphology was observed in C-32 melanoma cultures treated with juglone. Control C-32 cells showed fibroblastic shape, while juglone-treated C-32 cells showed spherical shape.
Effect of juglone on the viability and proliferation of melanoma cells and normal dermal fibroblasts in in vitro cultures
In all cell cultures of both normal fibroblasts and melanoma cells, the effect of juglone on the percentage of viable, metabolically active, and dying cells was observed (Fig. 3 and Fig. 4).
Fig. 2
CVDE and WST-1 assays were performed to assess the number of adherent, living cells in cell culture and to assess metabolic activity of cells. In CVDE assay, the crystal violet dye attached to the bottom of the culture vessel is bound by the DNA of living cells – the darker violet color indicates more living cells in the culture. In WST-1 assay, tetrazolium salts are converted to formazan by metabolically active living cells. The reaction leads to a color change (from pink to orange) in the culture medium
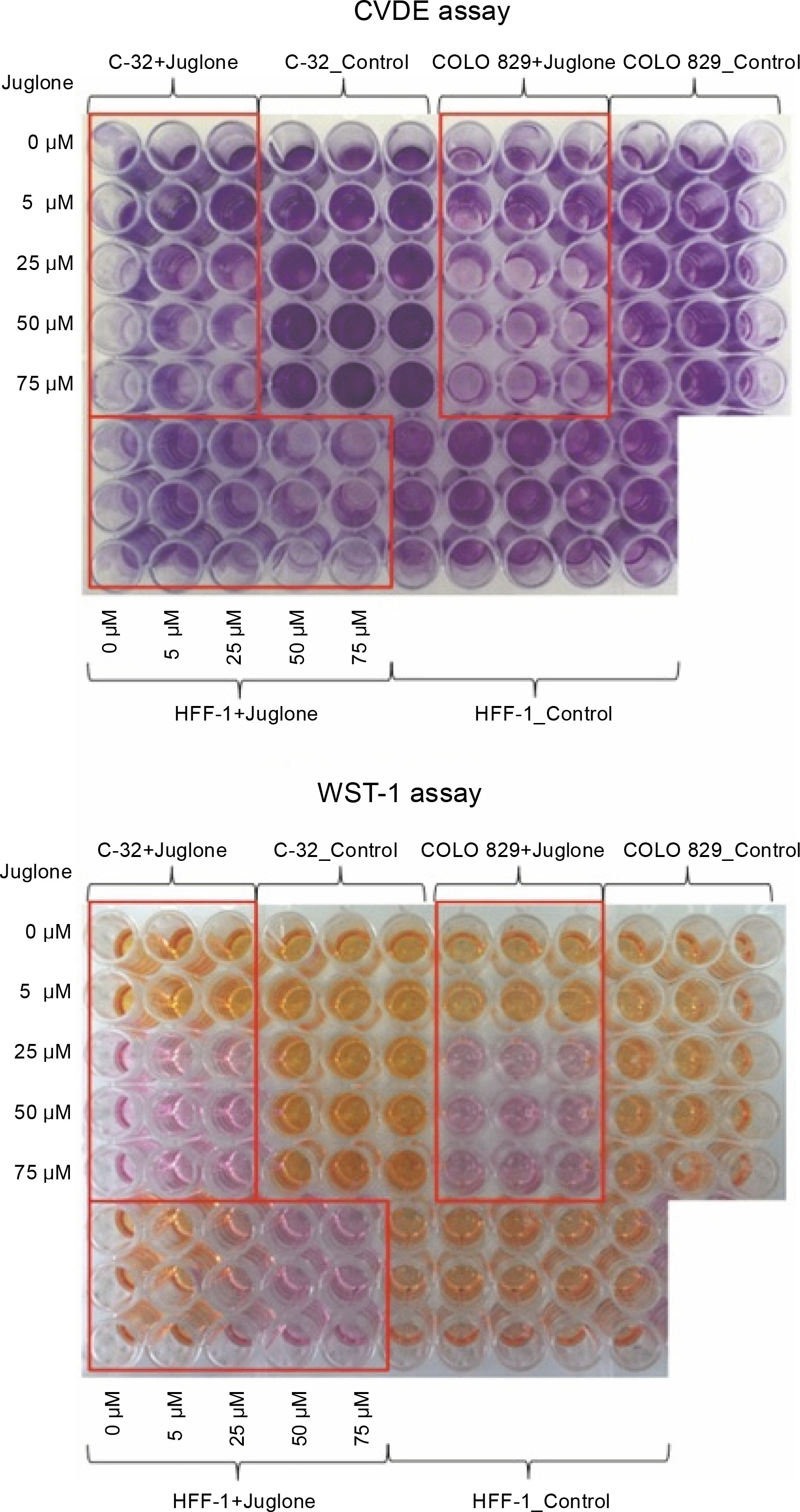
Fig. 3
The effect of different doses of juglone (0–75 μM) on the percentage of attached, viable cells (CVDE assay), on the metabolic activity of cells (WST-1 assay), and on the percentage of dying cells (LDH assay) in HFF-1, C-32, and COLO 829 in in vitro cultures. Statistical significance *P 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
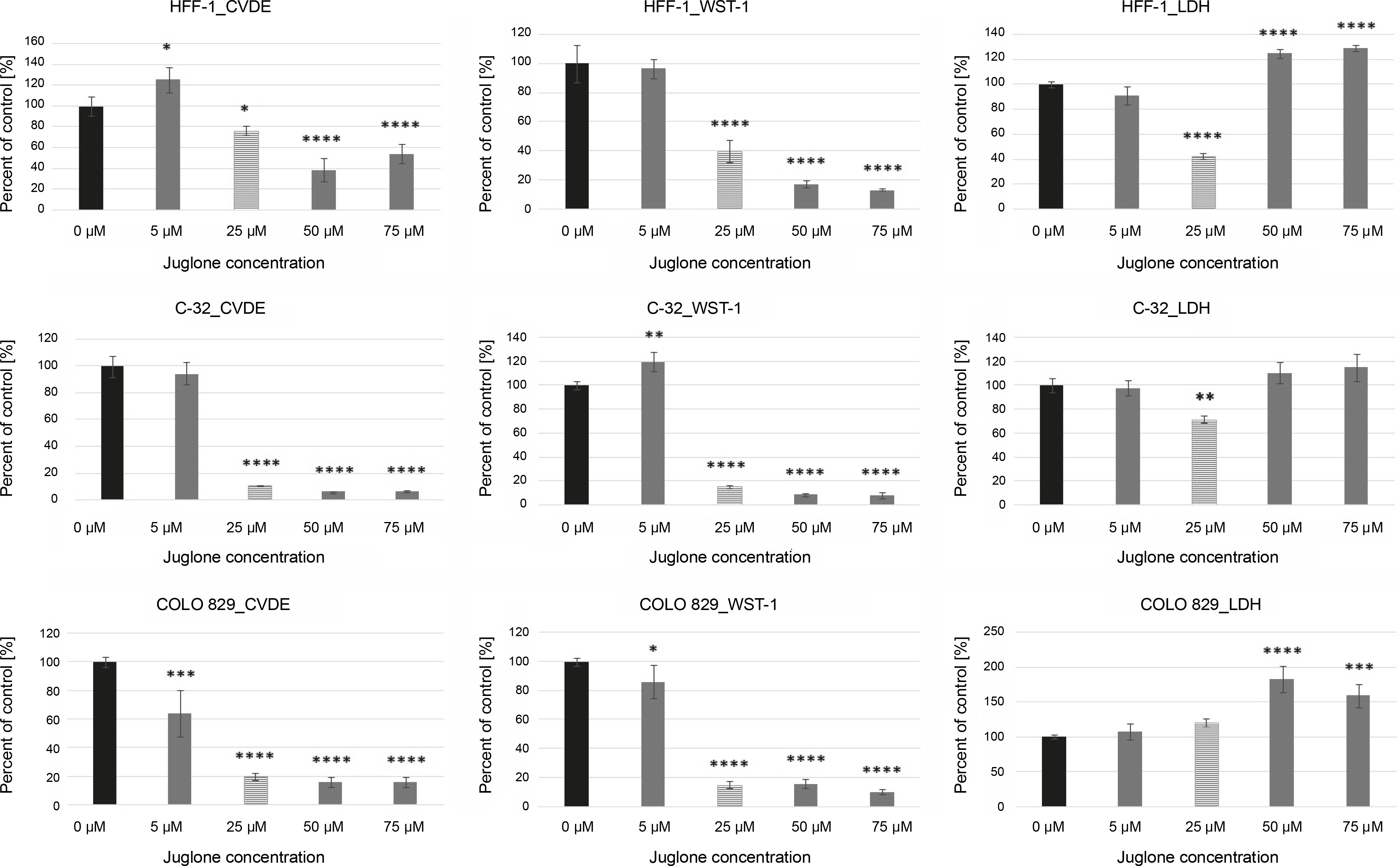
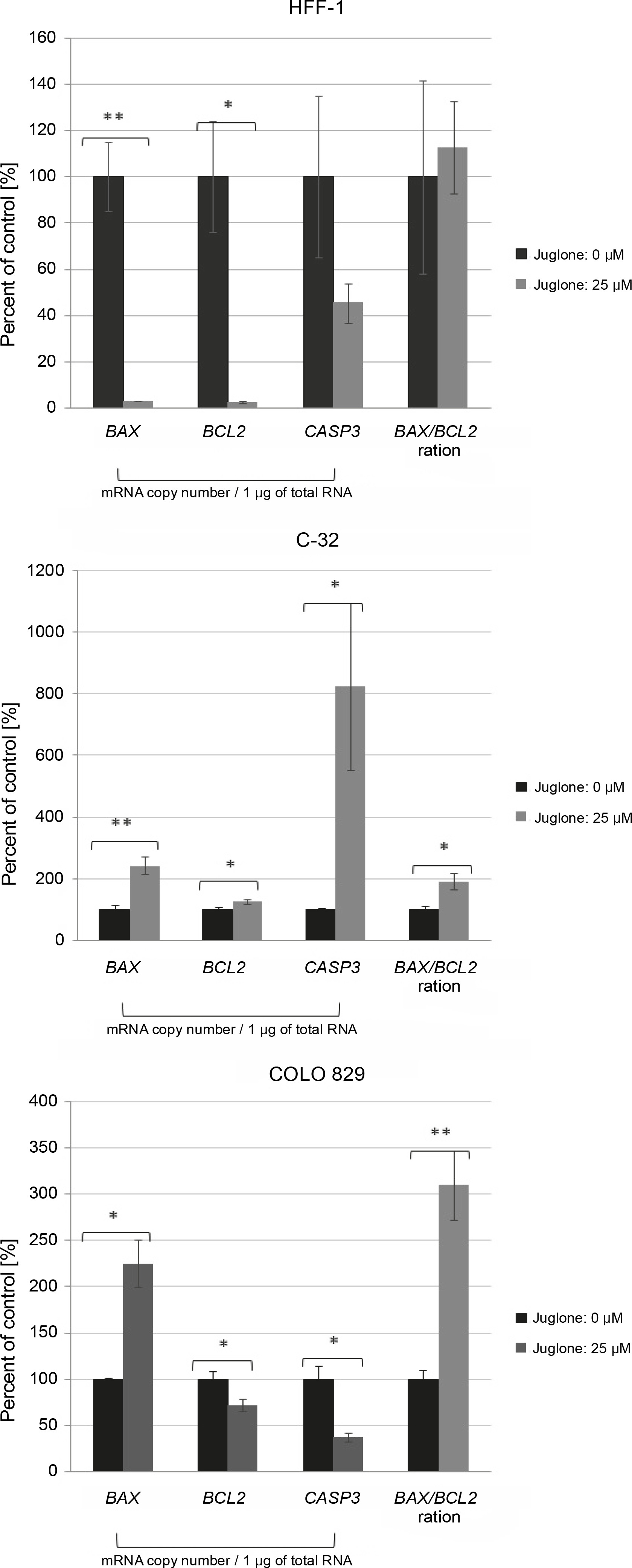
Fig. 4
The effect of juglone at 25 μM concentration on the BAX, BCL2, and caspase-3 mRNA level in human melanoma cells (C-32 and COLO 829) and normal dermal fibroblasts (HFF-1) in in vitro cultures as compared to control cultures (cells not treated with juglone). Statistical significance *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
As the concentration of juglone increased (from 50 to 75 μM), a decrease in the number of viable cells (CVDE test), a decrease in the metabolic activity of cells (WST-1 test), and an increase in the percentage of dead cells (LDH test) were observed.
The strongest effect of juglone was noted in both C-32 and COLO 829 melanoma cell lines. Juglone at 25 μM concentration caused: 1) a decrease in the number of living cells by approximately 90% in the culture of C-32 cells (****P < 0.0001) and by approximately 80% in COLO 829 cell culture (****P < 0.0001) and 2) a decrease in metabolic activity by approximately 85% in cultures of melanoma cells of both lines (C-32: ***P < 0.001; COLO 829: ****P < 0.0001). However, the effect of 25 μM juglone on the decrease (by approximately 28%) in the percentage of dying cells in C-32 cell culture (*P < 0.05) was noted; however, the percentage of cells dying in COLO 829 cultures exposed to juglone did not differ from that observed in the control. The effect of 25 μM juglone concentration on the increase in the percentage of dying cells in melanoma cultures was statistically significant only at the juglone dose of 50 μM and 75 μM for only COLO 829 cells. The 50 μM dose of juglone increased the percentage of dying COLO 829 cells by approximately 82% (****P < 0.0001), while the dose of 75 μM increased the percentage of dying cells by approximately 59% (***P < 0.001).
The effect of juglone was lower on HFF-1 normal dermal fibroblast cultures. Juglone at 25 μM concentration caused a decrease in the number of cells in the culture by approximately 23% (*P < 0.05) and a decrease in the metabolic activity of cells by approximately 60% (****P < 0.0001). An increase in the percentage of dying cells in HFF-1 culture cells treated with juglone was noted when its concentrations was 50 and 75 μM. The percentage of cell death increased in comparison to the control culture (not exposed to juglone) by approximately 25% and 29% for C-32 and COLO 829 melanoma cells, respectively (in both cases: ****P < 0.0001). Juglone at 25 μM concentration caused inhibition of cell proliferation in all the examined cell cultures of both normal and melanoma cells. Only higher juglone concentration caused induction of cell death in all cell cultures.
In summary, HFF-1 fibroblasts showed the lowest sensitivity to all tested juglone concentrations (5–75 μM) among all the tested cell lines. In contrast, COLO 829 melanoma cells were the most sensitive to the action of juglone. The lowest juglone concentration (5 μM) caused a statistically significant decrease in the number of cells by approximately 36% (CVDE, *P < 0.05) and a decrease in the metabolic activity of cells by approximately 14% (WST-1, *P < 0.05).
Quantitative assessment of the mRNA expression levels of BAX, BCL-2, and caspase 3 proteins
In cell cultures of both C-32 and COLO 829 melanoma cells and HFF-1 fibroblasts, a statistically significant effect of juglone at 25 μM concentration on the mRNA levels of BAX, CASP3, and BCL2 was noted. Each cell culture treated with juglone showed a different general expression profile of these three genes at the mRNA level (Fig. 4).
In cultures of normal dermal fibroblasts, the expression of all the analyzed genes (BAX, BCL2, and CASP3 ) was decreased at the mRNA level. However, the change in expression level was statistically significant only for the BAX and BCL2 (**P < 0.01, * P < 0.05), and their expression level was lower by approximately 98% as compared to that of the control culture (not treated with juglone).
In cell cultures of both C-32 and COLO 829 melanoma cells, following the treatment with juglone at 25 μM concentration, the transcriptional activity of the proapoptotic BAX was noted in relation to the control cultures. The BAX mRNA level was increased by approximately 143% in C-32 cell culture (**P < 0.01) and by approximately 126% in COLO 829 cell culture (*P < 0.05).
Melanoma cells of dissimilar cell lines showed a contrasting reaction with regard to the transcriptional activity of the BCL2 and CASP3 in response to juglone treatment. In C-32 melanoma cells, an increase in the mRNA expression level of both the BCL2 and CASP3 was noted.
The BCL2 mRNA level increased by approximately 26% (*P < 0.05.), and the CASP3 mRNA level was very significantly increased by approximately 725% (*P < 0.05) as compared to the control. In contrast, in COLO 829 cell cultures, a decrease in the mRNA expression level of both genes (BCL2 and CASP3 ) was noted. The mRNA expression level of the BCL2 and CASP3 decreased by approximately 28 and 62%, respectively (both *P < 0.05).
In C-32 and COLO 829 melanoma cell cultures treated with juglone at 25 μM concentration, a statistically significant increase in the BAX/BCL2 ratio (favoring the BAX mRNA transcripts) was noted (*P < 0.05 for both melanoma lines) as compared to that in control cultures. In the C-32 and COLO 829 cell cultures, the BAX/BCL2 ratio increased by approximately 92 and 210%, respectively, relative to the control culture (Fig. 4).
In HFF-1 fibroblasts treated with the same juglone concentration, there was no significant change in the BAX/BCL2 ratio (Fig. 4).
Discussion
Juglone, a component of walnut extract, is a promising chemical compound for use in oncotherapy. However, its clinical applications are still limited because of its poor solubility in aqueous solutions, poor absorption, and overall cytotoxicity to normal cells (Bayram et al., 2019). Yet, extracts from various varieties of walnut are used in traditional medicine to treat different diseases. The extracts have also been used in folk medicine for many years for treating cancer. The research conducted thus far has demonstrated the anticancer properties of juglone (Ji et al., 2011; Li et al., 2013; Fang et al., 2015; Delaviz et al., 2017; Wu et al., 2017; Bayram et al., 2018; Bayram et al., 2019).
The results of our present study showed a dose-dependent effect of juglone on the viability, proliferation, and death induction in C-32 and COLO 829 melanoma cells and HFF-1 fibroblasts in in vitro cultures. Juglone at 25 μM concentration resulted in a statistically significant decrease in cell viability (CVDE assay) and metabolic activity (WST-1 assay) in the cultures of both melanoma cell lines. The reduction in viable cell number was by approximately 90 and 80% for C-32 and COLO 829 cells, respectively (in both cases: ****P < 0.0001). Moreover, a lower effect of juglone was observed on HFF-1 fibroblasts (reduction in living cell number to approximately 23% (*P < 0.05). Metabolic activity was reduced in both melanoma cell lines by approximately 85% (in both cases: ****P < 0.0001), while it was reduced to approximately 60% in HFF-1 normal fibroblasts (****P < 0.0001).
The antiproliferative properties of juglone have also been confirmed in other studies on various types of cancer cells in vitro. For example, previous studies have demonstrated a strong dose-dependent and time-dependent influence of juglone on the growth of LS-174T colon adenocarcinoma cells (Li et al., 2013) and SGC-790 gastric cancer cells (Ji et al., 2011) and on the reduction of viability and inhibition of proliferation of SKOV3 ovarian cancer cells through blocking of cell cycle progression (transition from phase G1 to S) (Fang et al., 2015); juglone also strongly affected the inhibition of the proliferation of TCC-SUM and RT-4 bladder cancer cells (Bayram et al., 2018), MCF-7 breast cancer cells (Ji et al., 2016), and CCL-228-SW colon cancer cells (Bayram et al., 2019).
Caspases are intracellular proteolytic enzymes that belong to the family of conserved aspartate-specific cysteine proteases (Kopiński et al., 2014). These essential apoptotic enzymes are produced as inactive procaspases. They are activated by hydrolysis of the peptide bond at the site of aspartic acid, resulting in the activation of proteolytic caspase cascade. Because of their role in apoptosis, caspases are divided into two groups: initiating caspases (caspase 2, 8, 9, and 10) and executive caspases (caspase 3, 6, and 7). The initiating caspases initiate an apoptotic signal, while the executive caspases perform massive proteolysis leading to apoptosis. Caspases cleave a variety of substrates, including downstream caspases, nuclear proteins, plasma membrane proteins, and mitochondrial proteins, leading to cell death (Kopiński et al., 2014).
Caspase 3 is activated by both extracellular (mediated by death receptors) and intracellular (mitochondrial) pathways (Wang and Youle, 2009). PARP (poly (ADPribose) polymerase), a DNA repair enzyme, is probably the best characterized substrate for caspases. During apoptosis, PARP is cleaved into 24 and 85 kDa fragments corresponding to the N-terminal DNA binding domain and the C-terminal catalytic subunit, respectively. During apoptosis, PARP is selectively cleaved by several different caspases and particularly by caspase 3 (Wang and Youle, 2009).
Proteins belonging to the BCL2 family are the basic regulators involved in apoptosis. The most important proteins regulating apoptosis are the proapoptotic protein BAX and the antiapoptotic protein BCL2 (Li et al., 2013). The BAX protein is involved in the formation of permeability transition pores (PTPs), which leads to the induction of apoptosis. In contrast, the BCL2 protein plays the role of an inhibitor of PTP formation and prevents apoptosis (Li et al., 2013).
The increase in BAX protein level and decrease in BCL-2 protein level probably promote the formation of PTPs, inflow of Ca2+ ions into the mitochondrial matrix, disturbance of the osmotic pressure balance, and decrease in mitochondrial membrane potential (Ji et al., 2016). The formation of PTPs leads to a hypertonic environment within the mitochondria, which damages the outer mitochondrial membrane and causes release of apoptotic proteins such as cytochrome c, a component of the respiratory chain (Ji et al., 2016). Cytochrome c released into the cytoplasm of the cell leads to apoptosis by recruiting caspases (Wang and Youle, 2009; Ji et al., 2016). Apoptosis can be inhibited if the concentration of BCL2 is sufficient to form a complex with at least half of BAX (Li et al., 2013; Shamas-Din et al., 2013).
Our results showed a different expression pattern for the mRNA level of genes encoding the proapoptotic protein BAX and caspase 3 and the antiapoptotic protein BCL2 in melanoma cells and normal fibroblasts (C-32, COLO 829, and HFF-1) treated with juglone in in vitro cultures as compared to control cultures (not treated with juglone) (Fig. 4). In HFF-1 fibroblasts, juglone at 25 μM concentration caused a strong significant decrease in the BAX and BCL2 mRNA levels by approximately 98% (in both cases: *P < 0.05), but showed no statistically significant change in CASP3 mRNA level. In C-32 amelanotic melanoma cells, a significant increase in BAX (by approximately 143%, **P < 0.01), BCL2 (by approximately 26%, *P < 0.05), and CASP3 (by approximately 725%, *P < 0.05) mRNA level was noted. In COLO 829 melanotic melanoma cells, juglone at 25 μM concentration caused a statistically significant decrease in BCL2 (by approximately 28%, *P < 0.05) and CASP3 (by approximately 62%, *P < 0.05) mRNA level and a statistically significant increase in BAX mRNA level (by approximately 126%, *P < 0.05) (Fig. 4).
Finally, there was a statistically significant increase in the BAX/BCL2 ratio, in the favor of BAX mRNA transcripts, in both C-32 and COLO 829 melanoma cells treated with juglone (in both cases: **P < 0.01) in comparison to control culture (cells not treated with juglone) (Fig. 4). In contrast, there was no change in the BAX/BCL2 ratio in juglone-treated HFF-1 fibroblasts as compared to control. These results may suggest an effect of 25 μM juglone concentration on the promotion of cell death in C-32 and COLO 829 melanoma cells and a lower sensitivity of HFF-1 fibroblasts to this concentration of juglone.
Other in vitro studies have shown the effect of juglone on the induction of apoptosis through an intracellular (mitochondrial) pathway in A549 lung cancer, HL-60 human leukemia, and HeLa cervical carcinoma cells (Fang et al., 2015). These studies have shown that juglone induces apoptosis by increasing cytochrome c and caspase 9 protein levels released into the cytoplasm and increasing the BAX/BCL2 ratio. A gradual decrease in the procaspase 9 level was demonstrated with an increase in juglone dose-dependent caspase 3 activity in SKOV3 ovarian cancer cells (Fang et al., 2015). In MCF-7 breast cancer cells, LS-174T colon cancer line (Li et al., 2013), and SGC-7901 gastric cancer cells (Ji et al., 2011) treated with juglone, a decrease in BCL2 level and an increase in BAX level and caspase 3 activation were also noted (Ji et al., 2016).
Conclusions
The results of the present study indicate the potential antitumor activity of juglone on melanoma cells. The strength of juglone interaction and the final effect depended on the dose used and the type of cells. C-32 and COLO 829 melanoma cells were found to be more sensitive to lower doses of juglone as compared to HFF-1 fibroblasts. This effect is very desirable as it indicates the selective and inhibitory effect of juglone at a lower concentration range, wherein it targets melanoma cells, with a simultaneously lower impact on normal cells. However, it is necessary to conduct further detailed studies to determine the most desirable doses of juglone, which would spare normal cells and induce apoptosis in pathological cells. Moreover, it is necessary to investigate the exact mechanisms by which juglone exerts effects on various types of melanoma cells.
Sources of support and grant numbers: Medical University of Silesia, PCN-1-056/N/0/B.