






Introduction
Colorectal cancer (CRC) stands as one of the most prevalent malignancies globally and ranks among the primary causes of mortality in neoplastic diseases [1, 2]. Over the preceding decades, therapeutic approaches for CRC have undergone substantial transformations, resulting in enhanced overall survival rates for patients. Despite these achievements, a significant portion of new patients is diagnosed with metastatic disease, with approximately 15–30% of early-stage patients eventually relapsing [3]. Upon diagnosis of stage IV disease, comprehensive molecular testing becomes imperative prior to initiating first-line therapy. Specifically, evaluating microsatellite instability holds great significance because it can guide decisions regarding the application of immunotherapy [4]. Additionally, mutations in KRAS/NRAS or BRAF genes preclude the utilisation of targeted anti-epidermal growth factor receptor (anti-EGFR) therapy [4].
Hence, identification of molecular events is of utmost importance when initially treating a patient with metastatic CRC. Beyond the established RAS and BRAF mutations, recent data suggest that in subsequent lines of therapy, the application of other targeted agents may be considered, contingent upon the presence of specific mutations or gene amplifications. These encompass therapies targeting BRAF, anti-HER2 agents, as well as TRK inhibitors and RET fusion inhibitors [4, 5]. The extensively studied molecular diversity associated with metastatic CRC reveals that cancer cells tend to demonstrate spatial and longitudinal heterogeneity, acquiring mutations or undergoing changes in their mutational profile throughout the course of the disease and in response to the selective pressures exerted by administered therapy [6–8]. This phenomenon, whether arising from the delayed acquisition of mutations by cellular subclones or the gradual selection of initially imperceptible mutated subclones, suggests that anti-EGFR-based therapy holds the potential to significantly reduce the population of sensitive (wild-type) cells. This reduction, in turn, fosters the progressive predominance of resistant (mutant) clones until clinical evidence of disease progression becomes apparent. Subsequent treatments without anti-EGFR components may lead to the partial restoration of sensitive clones, thus establishing the foundation for the potential and documented efficacy of anti-EGFR rechallenge [6, 9].
Simultaneously, liquid biopsies have emerged as a secure and readily accessible alternative to conventional tumour tissue biopsies. This diagnostic approach involves analysing a sample from bodily fluids to identify primary circulating cancer cells, cell-free DNA, and other relevant molecules [10, 11]. Without the inherent drawbacks of tissue biopsy, they play a crucial role in capturing tumour mutational signature and offer valuable insights into the mechanisms underlying therapy resistance and molecular tumour evolution [12–14]. As shown by literature data, liquid biopsies find utility in scenarios where the re-administration of anti-EGFR therapy is a potential therapeutic strategy or when tissue samples are insufficient or unsuitable for the initial genetic evaluation of the tumour [9, 15, 16].
Among the various biomarkers integrated into liquid biopsy, circulating tumour DNA (ctDNA) has undergone extensive study in the context of CRC [17–20]. Circulating tumour DNA comprises fragments of DNA deriving from tumour cells that can be detected in bodily fluids and particularly in blood plasma. Those fragments have a length of 140–170 bp and a short half-life of approximately 2 hours [21]. Its biological characteristics allow for real-time monitoring of the molecular characteristics of the disease, providing information regarding response to therapy and the emergence of new clones timely for the oncologist to make the right therapeutic decisions. It has been shown that ctDNA levels correlate with tumour burden and patients with metastatic CRC have higher amounts of ctDNA, albeit being a small percentage of plasma total free DNA [11].
In order to reliably identify mutations using ctDNA, the application of techniques with very high sensitivity is a prerequisite. Polymerase chain reactions (PCR) such as BEAMing have reported a threshold of 0.01% [22]. Although they carry inherent drawbacks such as high cost, the inability to cover multiple hotspots, and lengthy protocols when compared with next-generation sequencing platforms, they remain a valid option for accurate detection of specific mutations [23]. BEAMing-Digital PCR (from beads, emulsion, amplification, magnetics), certified for use in in vitro diagnostics (CE-IVD), is the platform that can be used for extended RAS testing of the plasma. BEAMing-Digital PCR performs ctDNA analysis for a total of 34 mutations in KRAS exon 2 (codons 12, 13), exon 3 (codons 59, 61), exon 4 (codons 117, 146) and NRAS exon 2 (codons 12, 13), exon 3 (codons 59,61), exon 4 (codons 117, 146), with combination of digital PCR (dPCR) and flow cytometry, achieving a detection limit as low as 0.01% [22, 24].
To shed light upon the molecular mechanisms governing the evolution of CRC, as well as its response to therapy and the development of resistance, we opted to explore the mutational status of KRAS and NRAS in patients diagnosed with CRC in our institute. The aim of our study was to employ dPCR BEAMing for the detection of KRAS and NRAS mutations in individuals with CRC and evaluate the outcomes at various time points throughout the course of therapy.
Material and methods
Our observational study aimed to identify KRAS and NRAS mutations in patients with metastatic CRC treated at our clinic. Enrolment was open to patients with histologically confirmed CRC and stage IV disease, provided they furnished informed written consent. Blood samples were acquired from participants at various stages, including baseline upon disease diagnosis, during first-line therapy – specifically at the midpoint of therapy, and at first, second, and third disease progression (Fig. 1). These blood samples were stored and processed for ctDNA isolation and mutation identification using BEAMing. The observational study received approval from the Ethics Committee of the University Hospital of Ioannina (approval number 12401/30-4-2018). All enrolled patients underwent therapy in accordance with established guidelines and at the discretion of their treating physicians.
Fig. 1
Flowchart illustrating the collection of blood samples throughout the study, including samples taken at baseline upon disease diagnosis, as well as at first, second, and third disease progression stages
PD – progressive disease, tx – chemotherapy

From May 2018 to November 2021 a total of 99 patients were enrolled in the study, with the final participant enrolled on 10/11/2021. The last patient visit date occurred on 19/10/2022. Among the 99 enrolled patients 29 were female and 70 were male.
To assay the samples using dPCR, the BEAMing Digital PCR method was chosen, employing the OncoBEAM™ RAS CRC CE-IVD kit (Sysmex Inostics GmbH, Hamburg, Germany) following the manufacturer’s instructions. This assay detects 34 target mutations in codons 12, 13, 59, 61, 117, and 146 of the KRAS and NRAS oncogenes against a background of wild-type genomic DNA. Combining dPCR with flow cytometry, this assay achieves a detection limit as low as 0.01%. In the BEAMing process, each DNA molecule in a given plasma sample is compartmentalised in a ‘water-in-oil’ emulsion with a magnetic bead. The DNA molecules multiply, and thousands of identical DNA copies bind to the magnetic bead. Subsequently, DNA molecules are hybridised with tracers specific to the molecule type (derived from a wild-type or mutant allele, respectively). Qualitative and quantitative assessment is performed using flow cytometry within the initial population of DNA molecules [22].
Results
Among the 99 patients enrolled in the study, we finally acquired baseline blood samples from 71 individuals. Out of these 71 samples, 2 were deemed invalid upon BEAMing assay. Among the remaining 69 samples with valid results, 34 exhibited a wild-type status for the KRAS and NRAS genes (49.2%), while 35 carried mutations in either KRAS exon 2 codon 12, KRAS exon 2 codon 13, NRAS exon 2 codon 12, NRAS exon 2 codon 13, NRAS exon 3 codon 61, NRAS exon 4 codon 117, or a combination thereof (50.7%) (Fig. 2, 3).
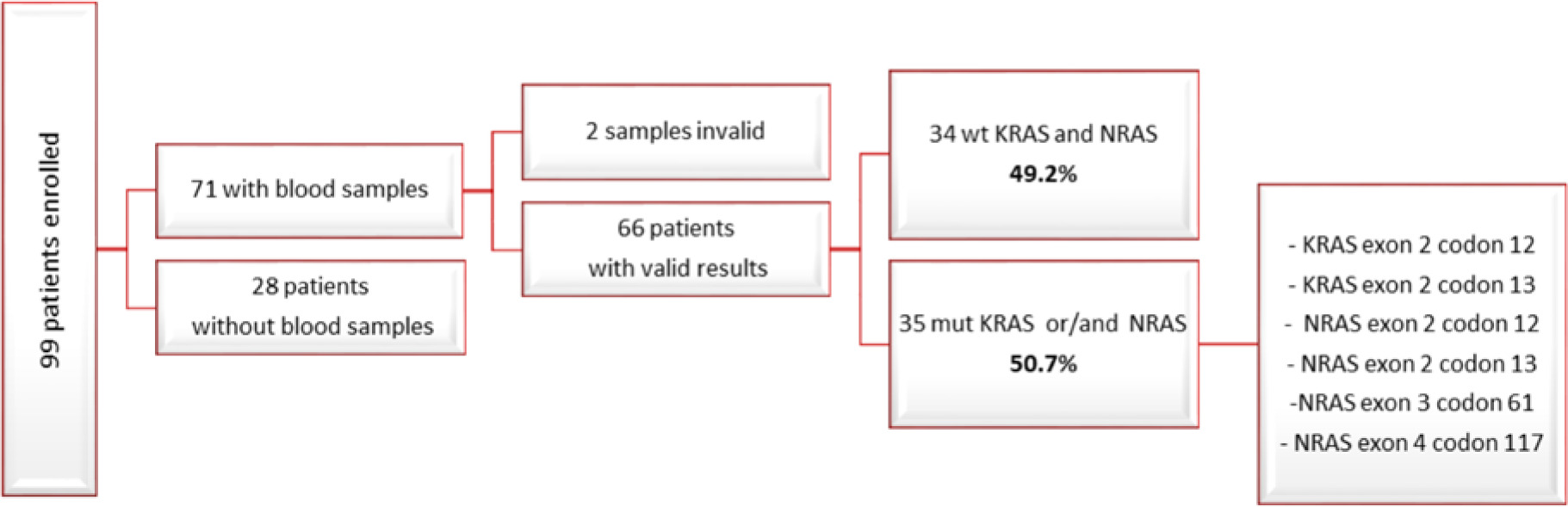
Fig. 3
Flowchart depicting the results obtained from first progressive disease blood samples
PD – progressive disease
Additionally, during the initial line of treatment, samples were collected from the same cohort at the midline of the first applied chemotherapy regimen. Out of the 71 patients assayed with BEAMing at baseline, 17 samples were collected during this phase. One sample yielded an invalid result after BEAMing, and another sample did not run at all. Notably, 14 samples retained the same mutations as observed at baseline. Furthermore, a mutational status change was observed in 3 specimens, specifically, 3 patients who initially carried RAS mutations were found to be wild type at midline testing.
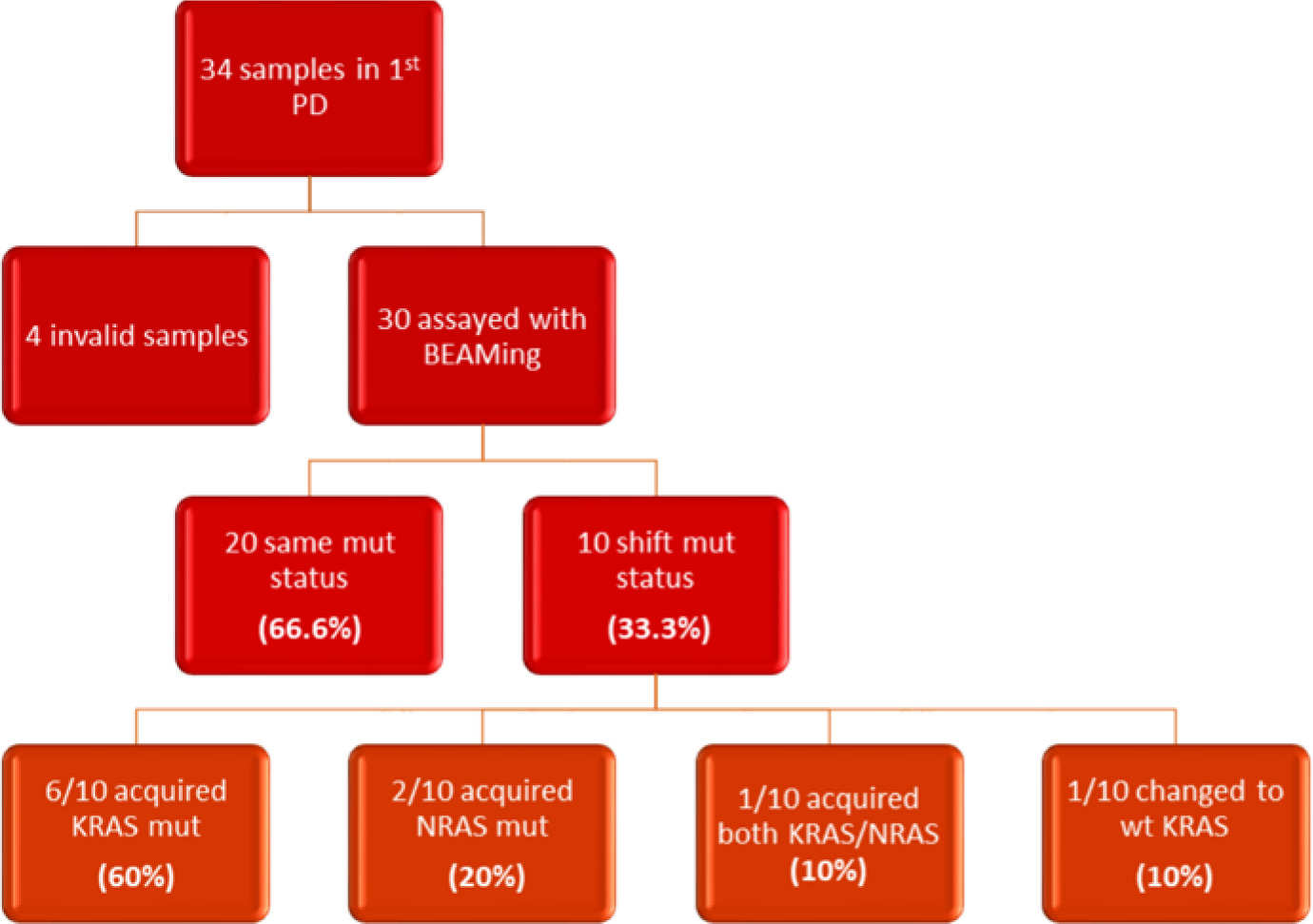
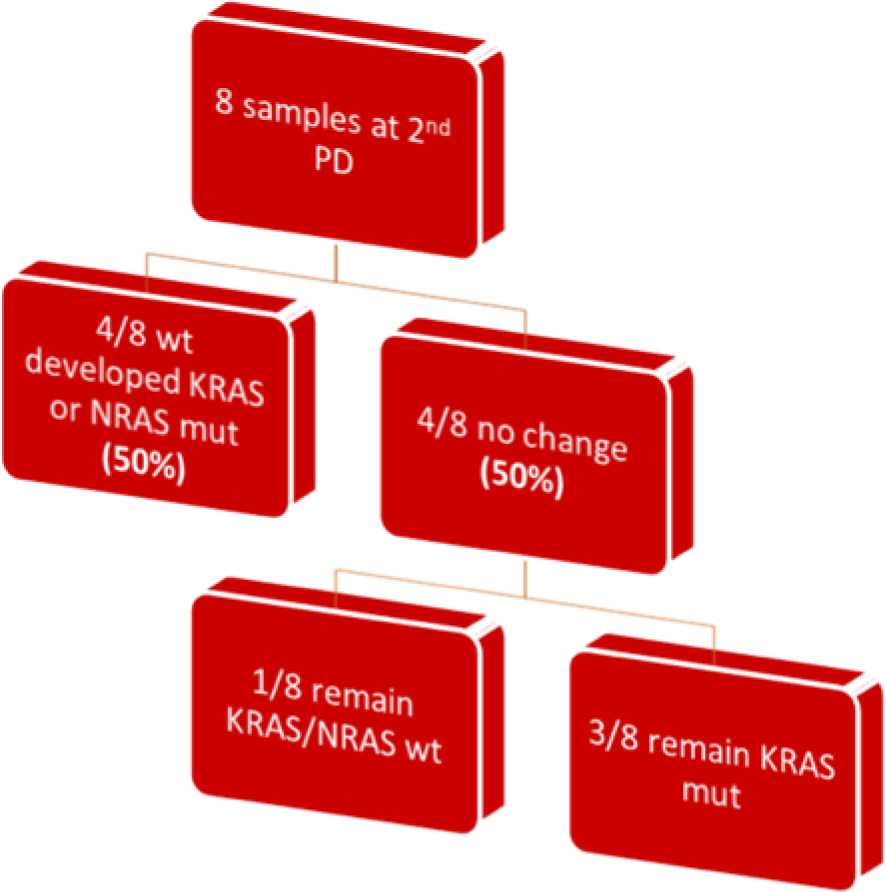
In the subsequent follow-up of the enrolled patients, 34 additional samples were collected at the point of disease progression after the first-line treatment. Of these 34 samples, one was considered inappropriate for further testing due to technical reasons, and 3 samples did not run at all. The remaining 30 samples were assayed with BEAMing. Among these, 20 samples maintained the same mutations at both baseline and disease progression (66.6%), while 10 samples exhibited a shift in mutational status (33.3%). Notably, 6 out of the 10 patients with mutational changes had acquired a KRAS mutation (60%), 2 had acquired an NRAS mutation (20%), and one patient acquired both KRAS and NRAS mutations at the first disease progression (10%). Finally, one patient, despite carrying a KRAS mutation at baseline, was found to be wild type at disease progression (10%) (Fig. 2, 3). Moreover, we successfully obtained 8 samples at the second disease progression of the enrolled patients. Among these, 4 exhibited no change in mutational status from baseline (50%). Three patients consistently carried a KRAS mutation that remained unchanged through both the first and second disease progressions, while one patient maintained a wild-type status at all time points. Additionally, 4 patients initially classified as wild type at baseline developed KRAS or NRAS mutations at the second disease progression (50%) (Fig. 4). Furthermore, 3 patients experienced a change in tumour mutational status between the first and second disease progression timepoints.
Discussion
Utilising dPCR BEAMing, we successfully identified and monitored the mutational status of KRAS and NRAS genes in patients with CRC enrolled in our observational study. The results align with those of similar studies in the literature because 66.6% had the same mutation status after the progression of disease, confirming the method’s high sensitivity and advocating for its extended applications in monitoring the mutational status of patients with metastatic CRC.
Furthermore, we identified one patient who, despite carrying a KRAS mutation at baseline, exhibited a wild-type status at disease progression. Similar to our study, Moati et al. observed a 22.2% conversion rate of RAS mutant plasma samples to RAS wild type [25]. Several studies have noted the frequent occurrence of this conversion in metastatic colorectal cancer (mCRC), ranging from 8 to 70% [25–29]. There are various potential reasons for this phenomenon. Firstly, investigations utilising liquid biopsy suggest that anti-angiogenic therapy may influence changes in RAS mutational status among mCRC patients. For instance, Sunakawa et al. documented a 76% conversion rate from RAS mutant to RAS wild type after several weeks of chemotherapy combined with bevacizumab [28], indicating a potential negative selection of RAS mutant clones during mCRC treatment. Another possible explanation for the non-detection of the mutation could be false negative results from liquid PCR tests. However, based on the literature and the sensitivity of these tests [30, 31], this explanation appears less likely to be the case. It is important to note that the limitation of the above data lies in the small number of individual studies, and conclusions should be drawn cautiously.
Approximately 40% of patients with mCRC exhibit KRAS and NRAS mutations [32], and their well-established negative predictive value is particularly relevant in the context of anti-EGFR monoclonal antibody therapies like cetuximab and panitumumab, as shown by extensive clinical trials [33–35]. Identification of RAS mutations prompts the exclusion of patients from anti-EGFR therapy.
Evidence indicates that discontinuation of anti-EGFR targeted therapy leads to the decline of RAS mutant clones within a few months, creating an opportunity for anti-EGFR rechallenge in a subset of metastatic patients, thereby offering additional clinical advantages [9, 36, 37]. Repeated tissue biopsies for RAS alteration identification are impractical. Early detection of emergent RAS mutant clones in patients initially classified as RAS wild type through baseline tissue testing allows for more vigilant monitoring, substantial therapy modifications, and a deeper understanding of evolving resistance.
Recent data suggest that the detection of changes in ctDNA reflecting the mutational status of these genes may present an opportunity for the successful re-application of anti-EGFR therapy in subsequent lines of treatment, thereby enhancing the empirical 8–20% response rate [38]. In prospective trials such as CHRONOS, CRICKET, and GEMCAD 17-01, patients with RAS/BRAF wild type were granted the opportunity for a therapeutic rechallenge if ctDNA testing revealed an absence of mutations in those genes. Those individuals who received personalised therapy based on their mutational status exhibited response rates ranging from 20 to 30% [39–41]. Therefore, monitoring guided by ctDNA facilitates the noninvasive molecular identification of patients within a subset of CRC who may exhibit a favourable response to anti-EGFR therapy. Ongoing large, randomised trials aim to further augment and refine these data [42].
Furthermore, liquid biopsy, employed clinically to assess existing gene alterations through ctDNA, has been investigated for its utility in early diagnosis, predicting recurrence or metastasis, and determining prognostic value in CRC patients [20, 43–45]. Published data support the use of ctDNA in mCRC [46–48], highlighting substantial concordance between tissue and plasma testing for KRAS and NRAS mutations [49]. However, the efficacy of ctDNA in detecting copy-number variations is confined to patients with elevated tumour content or extreme copy-number amplifications, and challenges persist in identifying copy losses in plasma [50]. In upcoming investigations, it is imperative to prioritise the evaluation of a comprehensive panel of mutations in ctDNA upon disease progression. This assessment plays a crucial role in identifying patients who may benefit from anti-EGFR monoclonal antibodies. This includes a thorough examination of MAPK alterations or ERBB2/MET amplification, acknowledged for their role in conferring resistance to EGFR blockade, albeit at a lower prevalence in mCRC [51]. Moreover, it is crucial to acknowledge that in patients with peritoneal carcinomatosis or brain metastasis, blood-based ctDNA may display limited sensitivity owing to blood-based barriers [52, 53]. Additionally, around 15% of patients may not exhibit detectable ctDNA, influenced by both tumour type and burden [50]. Ongoing investigations explore alternative methods for ctDNA detection in various body fluids, such as cerebrospinal fluid, ascites, and pleural effusion [50, 52, 53].
Conclusions
While the current observational study faces limitations, notably its non-randomised design and single-centre approach, profiling the tumour genomic landscape at the onset of metastatic disease and subsequent therapy timepoints has identified patients with newly evident mutations, particularly in RAS and RAF genes. These findings indicate potential resistance to anti-EGFR therapy. Conversely, a subgroup of patients no longer exhibited hotspot gene mutations post-chemotherapy, suggesting their eligibility for anti-EGFR targeted therapy. This information underscores the importance of an individualised approach to therapy selection, emphasising the application of personalised medicine in individuals with mCRC.