










Introduction
Coagulation factor VIII (antihemophilic factor A) is a glycoprotein synthesized mainly in hepatocytes, but also in kidneys, endothelial cells, and lymphatic tissue. It is one of the largest coagulation factors, with a molecular weight of 293 kDa. Factor VIII circulates in the bloodstream bound to von Willebrand factor in a non-covalent complex. This association was first described by Vehar et al. (1984) and Fang and Wang (2007). The active form of factor VIII (FVIIIa) functions as a nonenzymatic cofactor for the prothrombinase and tenase complexes in the intrinsic coagulation pathway. It enhances the activation of factor X by activating factor IX (FIXa) in the presence of phospholipids and calcium ions. The lack of this protein, which is characteristic of hemophilia type A, leads to a severe blood coagulation disorder. Factor VIII is composed of 2332 amino acids and consists of six domains, namely A1–A2–B–A3–C1–C2 (Kurachi and Davie, 1982).
In the bloodstream, proteolytic processes, specifically furin protease, cleave the factor VIII protein into two chains: a heavy chain (HC) of 200 kDa (A1–A2–B) and a light chain (LC) of 80 kDa (A3–C1–C2). These chains are connected by a covalent bond. Limited proteolysis of the B chain produces a heterogeneous population of active factor VIII forms with molecular weights ranging from 90 to 200 kDa. The smallest of these forms, composed of an HC of 90 kDa and an LC of 80 kDa, represents the active coagulation factor VIII. Notably, the resulting active form of the B domain lacks glycosylation sites, specifically amino acids Arg740 to Glu1649 (Stoilova-McPhie et al., 2014). Within the B domain, the N-terminal region’s Ser743 is connected to the C-terminal’s Glu1638, forming a specific site known as SQ comprising 14 amino acids (SFSQNPPVLKRHQR). This site is located between domains A2 and A3. The presence of this site enables intracellular cleavage of the 170 kDa single chain (SC) and the formation of the 80–90 kDa active complex. It is worth noting that the presence of specific amino acids at positions −1 and −4 relative to the Glu1649 site in SQ allows proteolytic cleavage by furin protease.
In its inactive or minimally active state, factor VIII serves as a cofactor in the blood coagulation process. Activation as a cofactor occurs only after proteolytic cleavage at the SQ site (Thompson, 2003; Ngo et al., 2008). Figure 1 illustrates the three-dimensional structure of B domain-deleted coagulation factor VIII. The gene responsible for encoding coagulation factor VIII is located on the X chromosome (Xq28) in humans. When a mutation occurs in this gene, it leads to a congenital bleeding disorder known as hemophilia A. It is important to note that this mutation primarily arises in male germ cells. The effect of this mutation is either the reduced or absent synthesis of factor VIII or the production of an abnormal protein (Hong et al., 2007). Treatment of bleedings in the course of hemophilia and related disorders consists of supplementation of missing coagulation factor i.e., its substitution (Marchesini et al., 2021).
Fig. 1
Three-dimensional structure of B domain-deleted coagulation factor VIII (Ngo et al., 2008); domains A1, A2, B, A3, C1, and C2 form coagulation factor VIII

Materials and methods
Bacterial media
Bacterial media used for research: LB medium (10 g/l bacto tryptone, 5 g/l yeast extract, 10 g/l NaCl) + 1.5 g/100 ml agar, liquid medium LB+10% PEG (10 g/l bacto tryptone, 10 g/l yeast extract, 5 g/l NaCl, 100 g/l PEG 6000, pH 6.1), for 1 l of medium 10 ml 1 M MgCl2 and 10 ml 1 M MgSO4 were added; liquid medium MM (minimal medium) (8 g/l K2HPO4, 2 g/l KH2PO4, 3 g/l NH4Cl, 0.15 g/l FeNH4, 3 g/l Na citrate, 0.9 g/l K2SO4, pH 7.4) after sterilization for 1 l of medium 10 ml 50% filtered glucose was added; liquid medium MM for quarter-technical scale culture (8 g/l K2HPO4, 2 g/l KH2PO4, 3 g/l NH4Cl, 3 g/l sodium citrate, 0.15 g/l FeNH4 citrate, 0.9 g/l K2SO4, 0.1 g/l yeast extract, pH 7.4), after sterilization for 1 l of medium 2 ml 1 M MgSO4, 10 ml 50% glucose, 0.1 ml 1 M CaCl2, pH 7.0 were added. Antibiotics used for research: ampicillin (Amp) 100 μg/ml, Chloramphenicol (Cm) 25 μg/ml, kanamycin (Kan) 100 μg/ml, and tetracycline (Tet) 10 μg/ml were purchased from Sigma.
DNA manipulation, transformation, and sequencing
DNA restriction, ligation, and gel electrophoresis were performed using standard techniques (Sambrook et al., 1989). All bacterial cell transformations with plasmid DNA were carried out through electroporation using 1 mm cuvettes (BTX) and the MicroPulserTM electroporator (BioRad). Electrocompetent E. coli cells were prepared following the standard techniques (Sambrook et al., 1989).
Plasmid DNA was isolated using the Plasmid Mini Isolation Kit (A&A Biotechnology), following the manufacturer’s instructions. Restriction enzymes, ligase, and DNA ladder were purchased from Roche, New England Biolabs, and Fermentas, respectively, and their usage followed the instructions provided by the manufacturers. The amplification of DNA was performed using the Biotools DNA Polymerase (Biotools B&M Labs) on a PTC-100 cycler from MJ Research, in accordance with the manufacturer’s guidelines. For protein molecular weight determination, prestained markers were used, namely the Multicolor Broad Range Protein Ladder (Thermo-Fisher) and the Full Range Rainbow (Amersham). DNA sequencing was conducted at Genomed SA (Warsaw, Poland), utilizing their commercially available sequencing facility. The primers used in this study were synthesized at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences. PCR products were separated on agarose gels and purified using the Gel-out Kit (A&A Biotechnology), following the manufacturer’s instructions. The Chromogenix Coamatic® Factor VIII Kit was purchased from DiaPharma Group, Inc.
Host strains
The laboratory strains of E. coli, namely NM522, DH5α, and E. coli Z, are derived from E. coli K12, a Gram-negative bacterium that belongs to the Enterobacteriaceae family. These strains exhibit characteristic colony morphology on TSA, forming round, flat, shiny colonies with smooth borders. In liquid LB medium, they grow uniformly as suspended cultures. Furthermore, these strains are capable of growth in liquid medium MM when supplemented with thiamine (2 μg/ml) and L-proline (at concentrations ranging from 50 to 200 μg/ml). The optimal growth conditions for these strains include a temperature of 37°C and a pH range of 6.5–7.5. Table 1 provides an overview of the host strains and their respective genotypes.
DNA origin
PCR amplification in this study utilized vectors that contained fragments of the factor VIII gene as a source of DNA. Table 2 provides information on clone IDs, collections, descriptions, and origins of the plasmids used in the experiment. For the synthesis of the factor VIII gene, primers and synthetic oligonucleotides were purchased from the Institute of Biochemistry and Biophysics, Polish Academy of Science.
Table 2
Clone IDs, collections, description, and origin of the plasmids
Plasmid vectors
The gene fragment of factor VIII was synthesized (GenScript USA Inc., US). Subsequently, it was cloned into the cloning vectors pBluescript II SK(−) and pUC19 using standard techniques (Sambrook et al., 1989). The plasmid vector pBluescript II (GenBank Accession No. ATCC 87047) carries the ColE1 replicon and the ampicillin resistance gene (ampR). On the other hand, plasmid pUC (GenBank Accession No. ATCC 37254) also contains the ColE1 replicon and the ampR gene. The plasmid pDB (Hartman and Mendelovitz, 1996) containing the deoP1P2 promoter (Valentin-Hansen, 1982; 1984), transcription terminator trpA (Pharmacia Biotech), and the tetracycline resistance gene (tetR ), was used for the construction of the prokaryotic expression system.
Construction of the gene encoding blood coagulation factor VIII without the B domain
To construct a synthetic gene for human coagulation factor VIII, we designed specific primers and synthetic oligonucleotides. These were utilized in the synthesis of factor VIII gene fragments. Given the large size of the blood coagulation factor VIII gene, which consists of 4320 base pairs, we devised three cloning vectors: pBluescript II SK(−) (referred to as pBluescript I), pUC19 (referred to as pUC II), and pUC19 (referred to as pUC III).
Figure 2 illustrates the cloning strategy employed for plasmid pBluescript I, highlighting the restriction sites and factor VIII gene fragments, including the gene fragment from the vector IOH10704 (Table 2), as well as gene fragments A, B, and C. Fragments A, B, and C were constructed using the DNA Assembly Method (Sambrook et al., 1989). This method involved the chemical synthesis of shorter fragments, followed by hybridization and ligation to form the longer molecule. Subsequently, PCR amplification with specific primers was performed to amplify the correct DNA strands (Nowak et al., 2015).
Fig. 2
Construction scheme for the cloning plasmid pBluescript I with restriction sites and fragments of the factor VIII gene: gene fragment from vector IOH10704, gene fragments A–C; site-directed mutagenesis was necessary in order to change restriction sites into the original sequence of blood coagulation factor VIII (Vector NTI AdvanceTM10)

Table 3 presents the primers used for the synthesis of the factor VIII gene segment referred to as Fragment 1. Figure 3 depicts the cloning plasmid pUC II, illustrating the restriction sites and fragments of the factor VIII gene. These include the gene fragments from the vectors F82385511 and F81629998 (Table 2), as well as gene fragments D, E, and F. Fragments D, E, and F were constructed using the DNA Assembly Method (Sambrook et al., 1989).
Table 3
Primers for the construction of the cloning plasmid pBluescript I
Fig. 3
Construction scheme for the cloning plasmid pUC II with restriction sites and fragments of the factor VIII gene: gene fragment from vector F82385511 and vector F81629998, gene fragments D–F; site-directed mutagenesis was necessary in order to change restriction sites into the original sequence of the blood coagulation factor VIII; AmpR – ampicillin resistance gene (Vector NTI AdvanceTM10)
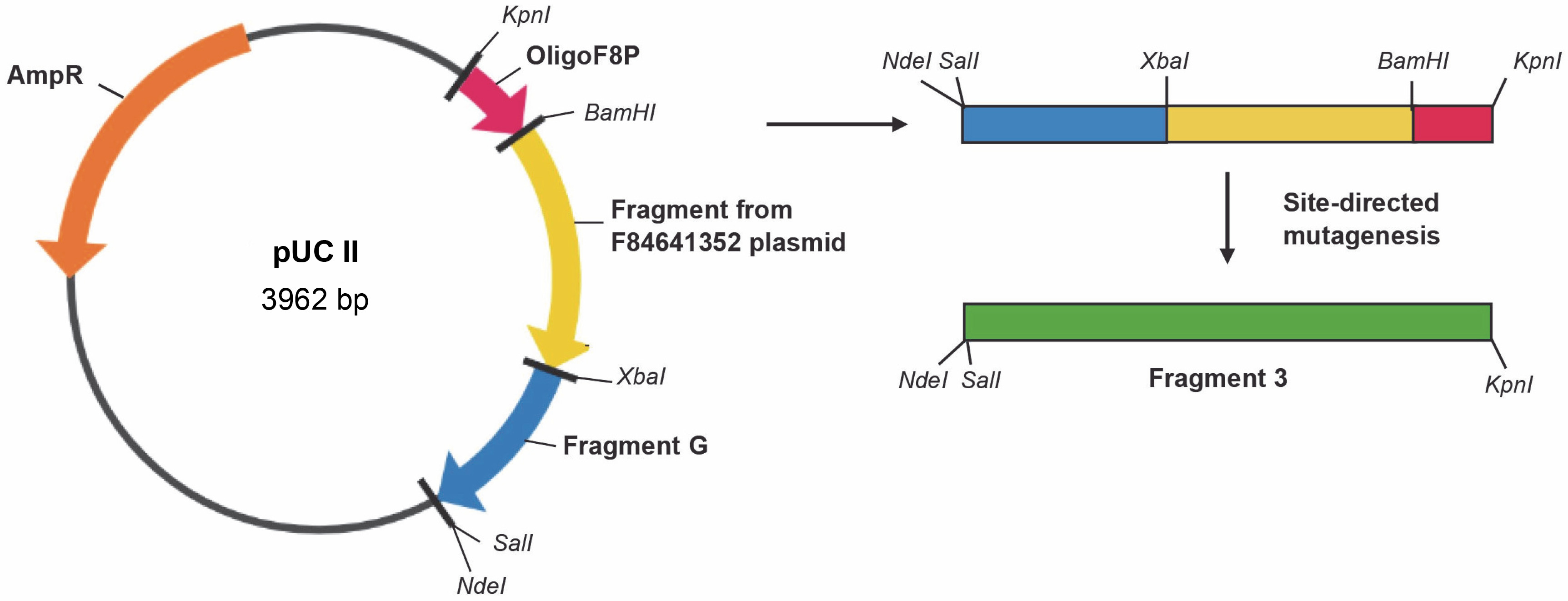
Table 4 provides the primers employed for the synthesis of the factor VIII gene segment known as Fragment 2. Figure 4 illustrates the cloning plasmid pUC III, displaying the restriction sites and fragments of the factor VIII gene. This includes the gene fragment from the vector F84641352 (Table 2), the synthetic oligonucleotide OligoF8P, and gene fragment G. Fragment G was constructed using the DNA Assembly Method (Sambrook et al., 1989). Table 5 presents the primers used for the synthesis of the factor VIII gene segment referred to as Fragment 3. The final plasmid, pBluescript + Fragment 1 + Fragment 2 + Fragment 3, was constructed by cloning Fragment 2 from the cloning vector pUC II and Fragment 3 from the cloning vector pUC III into the cloning vector pBluescript I + Fragment 1 (Fig. 5). All procedures throughout the study were conducted using standard techniques (Sambrook et al., 1989). To change restriction sites (SalI, ClaI ) into the original sequence of the blood coagulation factor VIII site-directed mutagenesis was performed using the QuikChange Site-Directed Kit (ThermoFisher). Finally, the sequence of the entire plasmid was confirmed, and the resulting construct was named pBluescriptFVIII.
Table 4
Primers for the construction of the cloning plasmid pUC II
Table 5
Primers for the construction of the cloning plasmid pUC II
Fig. 4
Construction scheme for the cloning plasmid pUC III with restriction sites and fragments of the factor VIII gene: gene fragment from vector F84641352, synthetic oligonucleotide oligoF8P, gene fragment G; site-directed mutagenesis was necessary in order to change restriction sites into the original sequence of the blood coagulation factor VIII; AmpR – ampicillin resistance gene (Vector NTI AdvanceTM10)

Fig. 5
Construction scheme for the cloning plasmid pBluescript + Fragment 1 + Fragment 2 + Fragment 3; site-directed mutagenesis was necessary in order to change restriction sites into the original sequence of the blood coagulation factor VIII; AmpR – ampicillin resistance gene, ori – origin of replication (Vector NTI AdvanceTM10)

Construction of the prokaryotic expression vector pDBFVIII
The pDBFVIII prokaryotic expression vector was constructed using the gene encoding factor VIII obtained from the pBluescriptFVIII plasmid and pDB expression vector. To introduce the restriction sites (SacII, XhoI ) into the pDB vector, we utilized the QuikChange Site-Directed Kit (ThermoFisher) for performing site-directed mutagenesis. The first mutagenesis reaction was performed to remove the XhoI restriction site located in the pDB transcription terminator trpA. Subsequently, the SacII and XhoI restriction sites were added. The resulting plasmid was then cleaved using SacII and XhoI enzymes. The 4320 bp fragment of the FVIII gene, obtained by digesting the corresponding region from the recombinant plasmid pBluescriptFVIII with the same enzymes, was inserted into the cleaved pDB vector in a clockwise orientation. To restore the original transcription terminator sequence trpA, another round of site-directed mutagenesis was performed using the QuikChange Site-Directed Kit from ThermoFisher. Finally, the complete plasmid sequence was verified to ensure its accuracy, and the resulting construct was designated as pDBFVIII (Fig. 6). The constructed prokaryotic expression plasmid carried a tetracycline resistance gene, conferring resistance to tetracycline antibiotics. Transcription initiation was controlled by the deoP1P2 promoter derived from E. coli.

Cell culture
The experimental work involved both laboratory-scale cultures in flasks and quarter-technical scale cultures in bioreactors. In the laboratory scale, the E. coli Z strain carrying the pDBFVIII plasmid derivatives was aerobically cultured at 37°C in LB or MM medium supplemented with tetracycline (100 μg/ml) until the optical density (OD) at 600 nm (OD600) reached a range of 0.5–1.5.
For the quarter-technical scale cultures, bacterial cells were obtained from a glycerol stock stored at −70°C. The E. coli Z strain containing the pDBFVIII plasmid with the factor VIII gene was initially grown for 12 h at 37−C in shaking flasks containing 50 ml of MM medium supplemented with a 50% glucose solution and tetracycline (100 μg/ml) until the OD600 reached approximately 1.0. These flasks (4 × 50 ml) served as the inoculum for the Bioflo 310 (New Brunswick Scientific) 7.5 l bench-top bioreactor with a culture volume of 4 l, as well as the Bioflo 415 (New Brunswick Scientific) 15 l bench-top bioreactor with an 8 l culture volume.
During the fermentation process, no antibiotics were added to the media. The cells were grown for 16–17 h at 37°C with an aeration rate of 5 l/min. To maintain glucose concentration within the range of 70–120 mg/dl during the exponential growth phase, a 50% glucose solution was added. The pH was controlled at around 7 throughout the entire run by adding 16% NH4OH solution. Once the OD600 reached approximately 25–37, the glucose feeding rate was automatically limited until the glucose concentration in the medium decreased to 0 g/dl. Subsequently, glucose feeding was controlled by the pH-stat method using a cascade system. Glucose was automatically added by the controller whenever the pH exceeded the set point of around pH 7 (with a dead band of 0.01). The glucose level was maintained within the concentration range of 30–40 mg/dl using the pH-stat approach. After induction, the culture was grown for an additional 4–7 h, reaching the stationary phase of growth.
Due to the minimal change in the culture volume (below 5%), the calculations for the batch fermentation were performed using a mathematical model based on Monod kinetics:
where X is the number or mass of cells (mass/volume), t is time, and μ is the specific growth rate constant (1/time).
Isolation, dissolution, and renaturation of inclusion bodies
The procedure for obtaining the recombinant blood coagulation factor VIII in the form of inclusion bodies involved several steps. Firstly, the E. coli Z cells expressing the protein were harvested through centrifugation at 9000 × g for 15 min at 4°C. The pelleted cells were then resuspended in a solution containing 100 mM Tris-HCl (pH 7.5), 500 mM NaCl, 10 mM EDTA, 0.35 mg/ml lysozyme, and 0.5 mM β-mercaptoethanol. Additionally, 1% PMSF (protease inhibitor) was added to prevent protein degradation. The bacterial suspension was gently mixed for 30 min at 20°C and subsequently lysed by sonication. Afterward, the lysate was centrifuged at 18000 × g for 20 min at 4°C.
The resulting pellet, containing the inclusion bodies, was washed twice with a solution of 50 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 1% Triton X-100 to remove bacterial proteins. Each wash step was followed by centrifugation at 18 000 × g for 20 min. Finally, the pellet was washed again with a buffer solution of 50 mM Tris-HCl (pH 7.5) and 500 mM NaCl.
To dissolve the inclusion bodies and extract the recombinant factor VIII, the pellet was suspended in a solution consisting of 8 M urea, 5 mM β-mercaptoethanol, and 50 mM phosphate buffer (pH 12). The suspension was gently stirred for 45 min at room temperature to facilitate the dissolution process. Subsequently, the suspension was centrifuged at 18000 × g for 15 min at 4° to remove any remaining insoluble debris. The factor VIII protein was then permitted to undergo folding over a subsequent 16 h period, accompanied by vigorous stirring and aeration at 8°C, within a renaturation buffer that consisted of 100 mM NaCl and 50 mM Tris (pH 10). The volume of the renaturation buffer utilized was 20 times greater than that of the renatured sample.
Protein determination by Western blot
To conduct SDS-PAGE electrophoresis, a volume of 55 μl of lysate obtained from E. coli, which had overexpressed coagulation factor VIII, was loaded onto the gel. Electrophoresis was carried out for 24 h to determine the protein size of 160 kD, using a 10% developer gel (Fig. 7). Subsequently, the proteins were transferred from the gel to a polyvinylidene difluoride membrane using a transfer apparatus. The membrane was then blocked using a blocking buffer.
Fig. 7
Expression level of the FVIII gene as revealed by SDS electrophoresis in a 12% polyacrylamide gel; 1 – protein marker Full Range Rainbow (Amersham), 2 – lysate of E. coli Z strain, 3 – (OD600 ~1.0) and 5 – (OD600 ~5.19) E. coli Z strain transformed with pDBFVIII plasmid, inclusion bodies isolated from LB + Tet medium, 4 – (OD600 ~1.0) and 6 – (OD600 ~4.50) E. coli Z transformed with pDBFVIII plasmid, inclusion bodies isolated from MM + Tet medium; SC – single chain, HC – heavy chain, LC – light chain

For the detection of coagulation factor VIII A2 (Pierce) domain, primary murine antibodies were diluted 1 : 1000 in the blocking buffer and applied to the membrane for incubation. Following a 1 h incubation period, excess unbound primary antibodies were removed by washing the membrane. In the subsequent step, horseradish peroxidase enzyme-conjugated secondary rabbit antimouse IgG antibodies (Pierce) were added. These secondary antibodies were diluted 1 : 1000 in the blocking buffer. Finally, a photographic plate was developed, revealing the analyzed coagulation factor VIII protein (Fig. 8).

Protein identification with 4800 Plus Maldi TOF/TOF
MALDI TOF/TOF spectra were obtained using a reflector mode on a 4800 Plus Analyzer from Applied Biosystems Inc. The matrix used for analysis was α-Cyano-4-hydroxy-cinnamic acid. External calibration was performed using a 4700 proteomics analyzer calibration mixture provided by Applied Biosystems. The acquired spectra were processed using Data Explorer Software, Version 4.9 from Applied Biosystems.
For peptide identification, the Mascot search engine from Matrix Science Inc. was utilized. The search was conducted against the Swiss-Prot and National Library of Medicine sequence databases. The protein bands corresponding to three forms of factor VIII (SC, HC, and LC) were identified based on the protein marker and confirmed by Western blot analysis. Following the procedure described by Sączyńska et al. (2018), the protein bands were excised from the gel, washed, and subjected to a reduction in the presence of dithiothreitol. Subsequently, alkylation with iodoacetamide was performed. The samples were then incubated with a trypsin buffer (Promega) at 37°C for 18 h. Before MS analysis, the samples were dried and dissolved in 10 μl of 0.1% trifluoroacetic acid.
Determination of factor VIII activity by chromogenic method
The activity level of factor VIII without domain B was determined using a chromogenic method, as there are differences in binding to phospholipids between native factor VIII and recombinant factor VIII without domain B. The Chromogenix Coamatic® Factor VIII Kit, following the manufacturer’s instructions from DiaPharma Group, Inc., was employed for the chromogenic FVIII activity assay.
The assay consisted of two stages (DiaPharma Group, Inc). In the first stage, the recombinant factor VIII without domain B, containing an unknown amount of functional FVIII, was added to a reaction mixture composed of thrombin, FIXa, FX, calcium, and phospholipid. This resulted in the rapid production of FVIIIa, which in turn, collaborated with FIXa to activate FX. The termination of the reaction allowed for the assumption that the production of FXa was proportional to the amount of functional FVIII present in the sample.
In the second stage of the assay, the measurement of FXa was carried out by utilizing a specific peptide nitroanilide substrate cleaved by FXa. This cleavage generated P-nitroaniline, resulting in the development of color. The absorbance of the color at 405 nm was measured using photometry. The intensity of the color produced was directly proportional to the quantity of functional FVIII present in the sample, based on a standard curve. To calibrate the standard curves of the factor VIII activity assays, the 7th International Standard Factor VIII from NIBSC (National Institute for Biological Standards and Control) was used as the factor VIII standard (Moser and Funk, 2014).
Results
Expression of factor VIII in E. coli prokaryotic system
To achieve overexpression of the factor VIII protein, we designed the prokaryotic expression vector pDB, which contained a synthetic factor VIII gene. This vector was then used for the transformation of the E. coli Z bacterial strain. In laboratory-scale experiments, we evaluated the expression of recombinant protein in two different media, namely LB and MM, supplemented with tetracycline at a concentration of 100 μg/ml. In the initial stage, the cultures were grown until they reached an optical density of OD600 ~1. In the case of cultures grown in MM+Tet medium, the expected OD600 ~1 was attained after 12 h, whereas in cultures grown in LB+Tet medium, the OD600 ~1 was achieved after 7.5 h.
In the subsequent stage, the cultures were further grown to attain maximum OD. After 24 h, the maximum OD600 was reached, measuring approximately OD600 ~4.50 for MM+Tet medium and OD600 ~5.19 for LB+Tet medium. Subsequently, all four cultures were subjected to centrifugation, and the inclusion bodies were isolated. These inclusion bodies were then separated using SDS electrophoresis on a 12% polyacrylamide gel, allowing us to evaluate the expression level of the FVIII gene. This evaluation took into account the type of medium used, the OD of the culture, and the amount of associated bacterial proteins (Fig. 7). Furthermore, the expression of the FVIII gene was confirmed through Western Blot analysis, using primary murine antibodies targeting the coagulation factor VIII A2 domain in the SC of FVIII (Fig. 8).
Protein identification with mass spectrometry
The confirmation of protein identity was achieved using MALDI-TOF/TOF mass spectrometry. The mass spectrometry analysis identified amino acid sequences based on the peptides obtained, aligning with the amino acid sequence of coagulation factor VIII. The identified amino acid sequences are depicted in Figure 9.
Fig. 9
The amino acid sequence of coagulation factor VIII with marked peptides; the amino acid sequences identified by mass spectrometry of peptides according to the amino acid sequence of coagulation factor VIII are marked with red color; SQN – proteolytic cleavage site for cutting the single chain form (SC) into the heavy chain (HC) and light chain (LC)

Bioreactor high cell density batch fermentation
To achieve higher cell densities, the cultivation process was scaled up. Initially, a 7.5 l bench-top bioreactor was utilized, with a culture volume of 4 l. Subsequently, the scale was further increased by employing a 15 l bench-top bioreactor, with a culture volume of 8 l.
Batch fermentation in 7.5 L bench-top bioreactor
The cultivation process in the 4 l culture volume bioreactor began with an initial OD600 (OD at 600 nm) of 0.13 and a glucose concentration of 90 mg/dl. Over the first 5 h, the bacterial culture utilized the glucose present in the medium for its growth. Subsequently, glucose was supplemented to maintain a concentration range of 60 mg/dl. The culture exhibited a particularly long lag phase lasting for 5 h. After this lag phase, the acceleration phase commenced, and after 11 h, the culture entered the exponential growth phase, which lasted for the following 7 h. The culture was considered complete when the OD reached its maximum value of 37. Several parameters of the cultivation process were monitored over time, including OD, real-time estimation of biomass (rx), dissolved oxygen (DO), and the rotational speed of the agitator. These parameters are presented graphically in the diagrams (Fig. 10).
Fig. 10
Batch bioreactor fermentation for E.coli Z strain with pDBFVIII plasmid; A, B – cell growth curve for 4 and 8 l, respectively; C, D – real-time estimation of biomass (rx) for 4 and 8 l, respectively; E, F – profiles of dissolved oxygen (DO) and stir speed for 4 and 8 l, respectively; R 2 – standard deviation

During the batch fermentation process in the 4 l culture volume, the rx was determined to be 1.67 × 1013 cells/h (1 OD600 = 8×108 cells/ml). The specific growth rate of the culture was calculated to be 0.2 l/h, with a corresponding generation time of 3.5 h. Following the fermentation of the E. coli Z strain with the pDBFVIII plasmid, approximately 160 g of wet biomass was obtained. From this biomass, 28 g of inclusion bodies were isolated, accounting for approximately 17.5% of the total biomass. The isolated inclusion bodies were subsequently dissolved and subjected to a renaturation process, as described in the Materials and Methods section.
Batch fermentation in 15 L bench-top bioreactor
In the second cell culture, the cultivation was conducted in an 8 l volume of LB medium. The initial OD600 was measured at 0.20, and the glucose concentration in the medium was 90 mg/dl. Similar to the previous culture, during the initial 3 h, the bacterial culture utilized the glucose from the medium for growth. Subsequently, glucose was added to maintain a concentration range of 70 mg/dl. The lag phase for this culture also lasted 5 h, similar to the smaller bioreactor. Following the lag phase, an acceleration phase of approximately 10 h was observed. This was followed by an 8 h exponential growth phase until the OD reached its maximum value of 25. The parameters of the cultivation process, including OD, rx, DO, and rotational speed of the agitator, were monitored and presented graphically in the diagrams (Fig. 10).
During the batch fermentation process in the 8 l culture volume, the rx was determined to be 3.13 × 1013 cells/h (1 OD600 = 8×108 cells/ml). The specific growth rate of the culture was calculated to be 0.351/h, with a corresponding generation time of 2.0 h. Following the fermentation of the E. coli Z strain with the pDBFVIII plasmid, approximately 238 g of wet biomass was obtained. From this biomass, 49.9 g of inclusion bodies were isolated, accounting for approximately 20.9% of the total biomass. The isolated inclusion bodies were subsequently dissolved and subjected to a renaturation process, following the protocol described in the Materials and Methods section. To analyze the protein expression levels of the FVIII gene, samples obtained from the batch fermentations and laboratory-scale culture were subjected to SDS electrophoresis in a 12% polyacrylamide gel (Fig. 11).
Fig. 11
Expression level of the FVIII gene as revealed by SDS electrophoresis in a 12% polyacrylamide gel; M – protein marker: Multicolor Broad Range Protein Ladder (ThermoFisher), 1 – batch fermentation in 8 l culture volume, 2 – batch fermentation in 4 l culture volume, 3 – laboratory scale culture; SC – single chain, HC – heavy chain, and LC – light chain

Based on the findings, it has been demonstrated that it is feasible to scale up the production process of the recombinant blood coagulation factor. The initial laboratory-scale culture was successfully followed by fermentation in a 4 l culture volume bioreactor, and eventually scaled up to fermentation in an 8 l culture volume bioreactor.
Determination of factor VIII activity by chromogenic method
To assess the activity of recombinant blood coagulation factor VIII, a chromogenic method was employed. This method measures the biological activity of factor VIII as a cofactor for the activation of factor X by active factor IX (factor IXa) in the presence of calcium ions and phospholipids. Before measuring the activity level of factor VIII, a standard curve was established. This involved preparing dilutions of the FVIII standard and measuring the corresponding absorbance values. Table 6 presents the dilutions used and the corresponding activity level values expressed in International Units (IU/ml).
Table 6
Dilutions of the FVIII factor standard according to activity level values in International Units IU/ml
Based on the dilutions, a standard curve was determined, described by the equation:
where x is the IU/ml activity level; y is the absorbance level A.
The relationship between the activity of the FVIII standard and the absorbance value is shown in Figure 12.
Fig. 12
Standard curve of the relationship between the activity of the FVIII standard and the absorbance value

After the renaturation process, the sample containing recombinant factor VIII protein at a concentration of 2.35 mg/ml was diluted by adding 25 μl of the sample into 2000 μl of the dilution solution. The absorbance of the diluted sample was then measured at a wavelength of 405 nm. Using the standard curve equation, the level of coagulation factor VIII activity was calculated based on the absorbance value. This activity measurement was performed for samples obtained from the laboratory scale, the 7.5 l bench-top bioreactor (4 l culture volume), and the 15 l bench-top bioreactor (8 l culture volume). Each sample was measured three times. The average activity level value from all measurements was determined to be 0.04 (Table 7). To assess the level of error, the standard deviation was calculated, resulting in a value of 0.000258736. This indicates an error range for the measured activity of approximately ± 0.0008.
Discussion
This study presents a method for obtaining recombinant factor VIII using a prokaryotic expression system. The protein’s antibody-binding properties were confirmed through Western blot analysis, and its activity was determined using the chromogenic method.
Hemophilia A is primarily treated with FVIII substitutive therapy. Early treatments involved lyophilized factor VIII concentrates derived from human plasma, which became available in the late 1960s (Pool et al., 1964). However, these concentrates were associated with serious side effects for patients. Due to the pooled plasma from multiple donors, they served as a source of hepatitis B virus and later hepatitis C virus since 1989 (Fletcher et al., 1989; Tobler et al., 1997; Rougemont, 2023). Moreover, in the early 1980s, 60–80% of hemophilia patients became infected with human immuno-deficiency virus (HIV) through these lyophilized concentrates (Curran et al., 1983; Evatt et al., 2006; Rougemont, 2023). A significant breakthrough in hemophilia treatment came with the discovery of the human factor IX and factor VIII genes in 1982 and 1984, respectively (Lusher et al., 1993). Shortly after, Wood et al. (1984) demonstrated that mammalian cells (BHK – baby hamster kidney cells and CHO – Chinese hamster ovarian cells) transfected with human factor VIII cDNA could successfully synthesize the factor. Recombinant factor VIII produced through genetic engineering technology became commercially available in the early 1990s (Bray et al., 1994).
In the 1990s, the availability of recombinant factor VIII raised expectations that it would replace human plasma-derived concentrates in hemophilia treatment (Coppola et al., 2010; Orlova et al., 2013; Lieuw, 2017). However, the adoption of recombinant factor VIII varies across countries. For example, Sweden and Ireland have reached 100% utilization of recombinant factor VIII among affected patients. In the United States, approximately 93% of patients receive recombinant factor VIII, while in Germany, the rate is around 72%. In Hungary, it is 60%, and in Poland, the utilization of recombinant factor VIII remains low at only 8% (World Federation of Hemophilia, 2021). Indeed, there have been significant advancements in the production of recombinant coagulation factor VIII and different generations of recombinant factor VIII pharmaceuticals have been developed (Kenneth et al., 2017). It is worth noting that all currently available recombinant factor VIII formulations have been produced using mammalian cell lines such as CHO, BHK, and human embryonic kidney cells (Lucas et al., 1996; Fussengger et al., 1999; Sandberg et al., 2012; Casademunt et al., 2012; Orlova et al., 2013; Valentino et al., 2014; Winge et al., 2015). However, breeding mammalian cells is time-consuming and very expensive, and the amount of the product obtained is usually low. In contrast to eukaryotic cells, a bacterial culture is inexpensive and allows the acquisition of large quantities of recombinant proteins in a short time (Rosano and Ceccarelli, 2014).
The findings presented in this paper highlight the successful production of an active form of recombinant blood coagulation factor VIII using a cost-effective prokaryotic expression system instead of the conventional eukaryotic-based system (Mazurkiewicz-Pisarek et al., 2016). This approach offers several advantages, including reduced manufacturing costs, shorter production time, improved product availability, and most importantly, elimination of the risk of infection associated with using plasma-derived products. The methods and results described in this study provide fundamental information for further research aimed at obtaining functional recombinant factor VIII in E. coli bacterial strains. While there are existing recombinant full-length factor VIII variants available for research or FDA approval, as well as studies reporting the production of recombinant factor VIII, to the authors’ knowledge, only two papers describe the production of recombinant factor VIII domains rather than the full-length FVIII factor. One of these papers reports on the production method of obtaining a recombinant A2 domain using insect cells and its activity confirmation via ELISA (Srivastawa et al., 2013), while another one describes the production of GST-tag or His-tag conjugated A1, A2, A2, and C domains using E. coli (Choi et al., 2015).
The advantage of our research lies in the successful production of an active, full-length recombinant factor VIII without the B domain and the use of any tags. This achievement opens up new possibilities for the development of a novel drug for the treatment of hemophilia A. Furthermore, future research in this field should focus on optimizing the production process to increase the activity of the obtained protein.
Conclusions
In this study, we present a novel method for obtaining recombinant FVIII factors using a prokaryotic expression system. Our experimental work successfully demonstrated the feasibility of constructing a synthetic gene for human coagulation factor VIII within the prokaryotic system. We described the process of constructing the prokaryotic expression vector, which involved cloning the synthetic gene encoding factor VIII. The expression of the vector was confirmed through mass spectrometry analysis.
Constructing such a large prokaryotic expression vector (8428 bp) and transforming it into the E. coli bacterial strain posed significant challenges. However, we were able to overcome these challenges and successfully obtain recombinant coagulation factor VIII in the form of inclusion bodies. We developed a method to isolate these inclusion bodies and determined the conditions for protein dissolution and renaturation while maintaining its activity.
As a result, we have devised effective scale-up strategies for a bioprocess intended to achieve the over-production of recombinant factor VIII on a quarter-technical scale. The successful development of a biotechnological method for producing recombinant blood coagulation factor VIII has validated our hypothesis that it is indeed feasible to obtain an active protein using an unexplored prokaryotic expression system.
In comparison to production in the eukaryotic system or by synthesis, the proposed biotechnological method of obtaining active recombinant factor VIII has the potential to significantly reduce production costs and increase production volume.