









Introduction
Fabry disease (FD), or as it is sometimes called Anderson-Fabry disease, is an X-linked metabolic error caused by various mutations in the α-galactosidase A gene (AGAL), which results in the accumulation of glycosphingolipids. It is one of the most prevalent lysosomal storage disorders, affecting many organs and systems; nevertheless, it is a rare disorder. Gastrointestinal symptoms are quite common in affected patients; therefore, it is important for gastroenterologists to keep it in mind as a differential diagnosis for especially challenging patients. The following review will focus on the diagnosis, gastrointestinal signs and symptoms, and possible forms of treatment.
Epidemiology and genetics
The worldwide incidence of classic FD is estimated at 1 : 40,000 to 1 : 117,000 live male births [1]; however, newborn screening has shown that the disease is much more common and can affect 1 : 3100 male infants [2]. These statistics reflect the old belief that women are only carriers – women are also affected with an estimated prevalence of 1 : 6000 to 1 : 40,000, although the exact numbers remains unknown [3]. The disease is underdiagnosed due to its rarity, non-specific symptoms, and atypical or late-onset presentation.
The deficiency of lysosomal α-galactosidase A gene (AGAL) causes the accumulation of glycolipids such as globotriaosylceramide (Gb3) and globotriaosylsphingosine (LysoGb3) [4]. LysoGb3 has many negative effects, including profibrotic, proinflammatory, and cytotoxic [5]. As Gb3 accumulates in various cells and organs (especially vascular endothelium, vascular smooth muscle cells, and pericytes) it leads to a whole spectrum of signs and symptoms.
Because the disease is X-linked, affected males are usually more symptomatic than females and more often have the so-called classic variant. They pass the defective gene to all their daughters and none of their sons. Females, on the other hand, are heterozygotes and therefore have a 50% chance of passing the gene to both daughters and sons. The course of the disease is also variable in women due to random X-chromosome inactivation and ranges from asymptomatic to a classic picture previously thought to occur only in males [6].
Until now, up to a thousand mutations in the AGAL gene have been identified [7]. Due to many reasons (e.g. epigenetic and environmental factors), it is extremely hard to establish a genotype-phenotype correlation. In general, mutations that result in null AGAL activity cause the classic variant, while if there is residual AGAL activity, atypical or late-onset disease is present [8].
Signs and symptoms
Classic disease
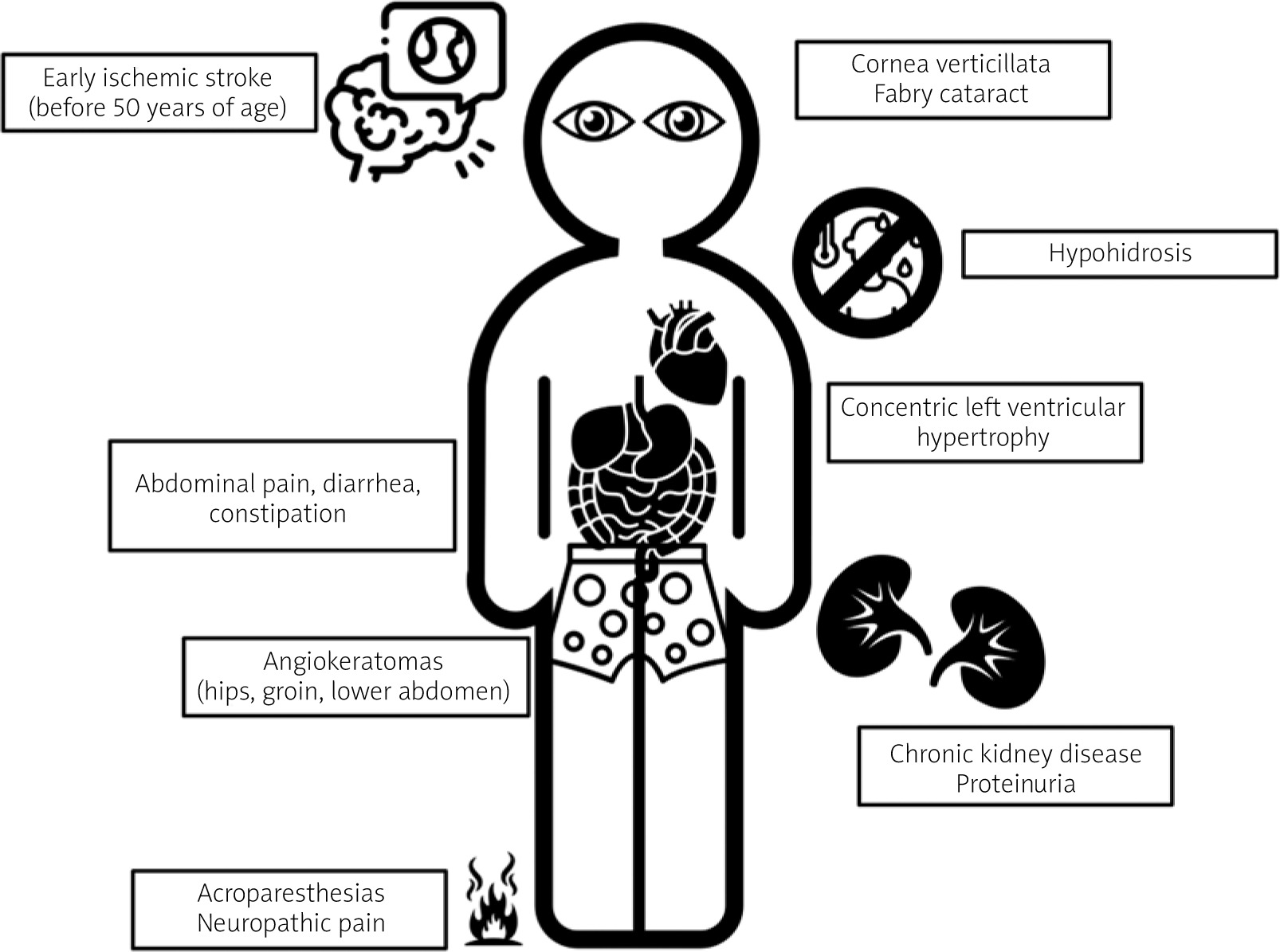
It usually occurs in males, although has been reported in some females. Symptoms appear in a predictable order starting from childhood to adulthood and becoming most prominent in the fourth decade of life (most symptoms are presented in Figure 1). The first symptom is very often severe neuropathic limb pain (acroparesthesias), which can be precipitated by extreme temperatures or stress. Characteristic telangiectasias and angiokeratomas (especially in the hips, groin, and lower abdomen) usually develop before the 20th birthday. Heat, cold and exercise intolerance, and hypohidrosis are almost always present. Corneal opacities and “Fabry cataract” are relatively common. Unexplained kidney function impairment and/or proteinuria occur in more than 80% of patients and may lead to end-stage kidney disease [9]. Cardiovascular symptoms include heart failure caused by left ventricular hypertrophy (LVH), myocardial fibrosis, coronary artery disease, aortic and mitral valve abnormalities, aortic root dilation, conduction abnormalities, and arrythmias [10]. Transient ischaemic attacks and ischaemic strokes occur before the age of 50 years. In addition, other neurological symptoms may be present such as blindness, sudden deafness, cognitive problems, and depression [11]. Heterozygous females may exhibit only some or sometimes all of the symptoms and signs of Fabry disease. Usually, symptoms appear at a later age, although the median age of onset among females is 13 years. In a large registry of more than 1000 women with FD, almost 70% had signs of disease [12].
Atypical (later-onset) variants
Those patients usually present later in life (third to seventh decades of life). There is also the cardiac variant, in which LVH, hypertrophic cardiomyopathy, conduction abnormalities, and arrhythmias predominate. It may be responsible for up to 4% of cases of otherwise unexplained hypertrophic cardiomyopathy [10]. The renal variant involves patients with disease limited to the kidney. The estimated FD prevalence in patients on dialysis is around 0.2–0.5% [13].
Gastrointestinal symptoms
Some authors report that gastrointestinal manifestations are present in as many as 70% of patients with FD. The most mentioned symptoms are nausea and vomiting, abdominal pain, and diarrhoea or constipation [14]. They are probably caused by Gb3 deposition in the autonomic nervous system of the bowel and in mesenteric blood vessels. This results in neuropathy, vasculopathy, and inflammation; also, the rates of gastroparesis and small intestinal bowel overgrowth (SIBO) are higher in affected patients [15, 16].
New studies estimate the prevalence of GI symptoms to be around 50% [17]. Female patients report symptoms more often than male patients, and children report symptoms more frequently than adults. The most common gastrointestinal symptom is abdominal pain, with similar distribution between males and females and median age of onset of 14 years. Diarrhoea is slightly less common and observed more frequently in men. Nausea and vomiting affect around 10% of patients. Bloating is also frequently observed. Other, less common symptoms included haemorrhoids, gastritis, or pancreatitis. The mean body mass index is similar in patients with and without gastrointestinal symptoms. Most commonly a combination of symptoms exists, usually abdominal pain and diarrhoea. The quality of life is significantly impaired in those who report gastrointestinal symptoms [14]. Overall, the clinical picture of Fabry disease is very similar to diarrhoea-predominant irritable bowel syndrome, and misdiagnosis is very common – the correct diagnosis is often obtained after as many as 20 years, and patients with predominant gastrointestinal symptoms are diagnosed with many diseases; in a South American cohort one of the most frequent was food intoxication [18].
It is important to precisely assess GI symptoms in Fabry disease. Many instruments can be used, but a specific FABry disease Patient-Reported Outcome-GastroIntestinal (FABPRO-GI) questionnaire is available. Two versions exist (24-h and a 7-day) for use in clinical trials and real-world settings, respectively. It includes questions on bloating, stomach pain, cramping, nausea, acid reflux, heartburn, indigestion, diarrhoea, constipation, and bowel movements [19].
Diagnosis
The diagnostic algorithm is quite simple and gender specific. The available tests are as follows:
All 3 tests can be measured easily by dried blood spot (DBS) testing – a form of biosampling where blood samples are blotted and dried on filter paper; therefore, only a small amount of whole blood is necessary.
Male patients can start with AGAL activity: if it is below 3%, the diagnosis is established, and mutation analysis should follow to guide treatment. If it is between 3% and 35%, the diagnosis is likely, and mutation analysis is needed to confirm the diagnosis. If it is above 35%, FD is excluded. Female patients need mutation analysis for diagnosis [20]. Plasma LysoGb3 levels, if elevated, may help confirm the diagnosis of FD among patients with gene variants of unknown significance, and they can also monitor the effects of disease-specific therapy [21].
Treatment
The treatment should consist of both supportive care and disease-specific therapy. That includes enzyme replacement therapy (ERT) and migalastat.
ERT replaces the deficient enzyme – AGAL – but requires biweekly injections. Three formulations of recombinant AGAL are currently available (although availability and reimbursement may differ between countries): agalsidase α, agalsidase β, and pegunigalsidase α (pegylated recombinant AGAL). The efficacy of agalsidase α and β formulations is presumed to be similar, but it has never been directly compared in randomized control trials. Pegunigalsidase α was noninferior to agalsidase β based on kidney-disease-specific endpoints. It also had a favourable tolerability and immunogenicity profile, with lower rates of treatment associated side effects [22].
Migalastat is an oral chaperone that binds to and stabilizes some mutant forms of AGAL; therefore, it is effective only in certain mutations and should not be used in patients with CKD stage 4–5 [23].
Expert guidelines are available on when to start disease-specific therapy, but clinical practice may vary between countries. Whenever possible, treatment should be provided by a centre with expertise in treating FD. Generally, classic disease, symptomatic female heterozygotes, and patients with later-onset variants should be treated with disease-specific therapy [24]. The proven benefits of disease-specific therapy are as follows:
lower tissue levels of LysoGb3,
lower rate of glomerular filtration rate (GFR) decline,
lower number of patients reaching end-stage kidney disease (observational data),
lower left ventricular wall thickness and stabilization or improvement of left ventricular mass,
less neuropathic pain,
stroke prevention (observational data),
New treatment options are being studied, the most promising of which is gene therapy with isaralgagene civaparvovec, which was recently granted fast track designation by the Federal Drug Agency (FDA) [28].
Treatment of gastrointestinal symptoms
According to European recommendations, the presence of GI symptoms that are not successfully treated with symptomatic therapy is an indication for initiation of disease-specific therapy (class IIA if < 16 years of age; class IIB if > 16 years of age) [29]. Abdominal pain is reduced in about 10% of patients at 12 months after initiation of ERT. Changes are similar in male and female patients. Similarly, the prevalence of diarrhoea is reduced by around 8% after ERT, with improvement being more pronounced among male patients. Treatment with ERT therefore has positive effects on GI symptoms but does not alleviate them completely. However, it might play a preventative role in the development of those symptoms. For instance, in a randomized controlled trial, no child without abdominal pain at baseline developed this symptom during 24 months of ERT [14]. There are scarce data on migalastat; however, it resulted in a significant reduction of GI symptoms after 24 months of treatment in naïve patients, and in patients previously treated with ERT it led to an improvement in diarrhoea [30, 31].
Because GI symptoms persist despite disease specific therapy, supportive treatment is needed. A few strategies have been prospectively tested, and they will be briefly described.
There have been studies exploring the relationship between LysoGb3 and the human microbiome. In an experimental study of an in vitro biofilm, it modified the composition of microbiome (promoting B. fragilis development and reducing Bifidobacterium, Akkermansia, and Lactobacillus) and altered the short-chain fatty-acid profile of this microbiota, leading to decreased production of butyrate [32]. Since FODMAPs (Fermentable Oligosaccharides, Disaccharides, Monosaccharides, and Polyols) can be involved in GI manifestations and dysbiosis; in other patients it was postulated that a low-FODMAP diet might be effective in Fabry disease. A small prospective study in FD patients with GI symptoms evaluated the effects of this diet. Although only around 50% of the tested population completed the intervention, this dietetic protocol seemed to have a positive impact on intestinal symptoms. It significantly improved indigestion, diarrhoea, and constipation [33]. More studies are needed, but eliminating food high in FODMAPs is the only dietary intervention with proven benefit and therefore should be recommended to willing patients.
Dietary supplements containing orally delivered AGAL also seem to be beneficial. A small study reported beneficial effects on abdominal pain and diarrhoea after a daily dose of 1800 U AGAL for 90 to 180 days [34]. It is postulated that it may work locally by potentially depleting intestinal LysoGb3, alleviating dysbiosis and local inflammation.
Based on the above-mentioned studies, comprehensive treatment plans should include ERT or chaperone therapy, nutritional support (low-FODMAP diet), and orally delivered and locally acting AGAL supplements [35].
Many other strategies such as the Okinawa diet, probiotics, proton pump inhibitors, metoclopramide, butyrate, etc. have been proposed, but they have not been prospectively studied. Until now, other treatments may be tried on an individual basis, but they remain unproven [36].
Summary
Although Fabry disease is rare, gastrointestinal symptoms are common in affected patients. An average patient sees at least 10 specialists [37] before receiving a diagnosis; therefore, it is important to remember this entity, especially in complex patients. The treatment consists of disease-specific therapy, which should be provided by centres with expertise, but also optimal supportive care, which is especially important in controlling gastrointestinal symptoms.