










Introduction
African swine fever (ASF) continues to have a detrimental impact on the global pig industry, particularly in Asia–world’s largest pig producer (Kedkovid et al., 2020). Despite efforts to control the disease through measures like culling infected pigs and implementing strict sanitary and monitoring policies, the lack of available vaccines for commercial use has hindered any significant progress. In addition, current management approaches and strategies have not effectively mitigated the huge socio-economic losses caused by ASF (Blome et al., 2020).
According to the World Organization for Animal Health (WOAH) report in December 2022, ASF has affected 45 countries across five regions in the world since January 2020. Total animal losses have exceeded two million, wherein over 1 191 000 pigs and 37 000 wild boars were affected (WOAH, 2022).
During the initial waves of outbreaks, small-to-medium pig farms with weak biosecurity measures have been identified as a crucial contributing factor (Kedkovid et al., 2020). This is because small backyard farms, despite their size, can have collective supply strain which has more significant impact as compared to larger pig farms (Cooper et al., 2022). In countries like China and Vietnam, small farms constitute approximately 80% of all pig farms. The proximity of small and larger farms can potentially facilitate the rapid spread of ASFV in Asian farms (Kedkovid et al., 2020).
ASFV strains circulating in Asia and Europe, particularly the highly virulent genotype II strains, are known to cause acute and peracute diseases within a short span of 7–10 days, often resulting in a mortality rate of up to 100%. Common symptoms of ASF include loss of appetite, high fever, gastrointestinal and respiratory manifestations, ataxia, cyanosis, and rapid-onset death. Clinical signs may vary depending on the specific strain of the virus (Blome et al., 2020).
ASFV is a DNA arbovirus belonging to the Asfarviridae family and genus Asfivirus. The viral genome of ASFV encodes approximately 150–200 viral proteins, including 68 structural and 100 nonstructural proteins which further highlights its complexity (Wang et al., 2021). Its complication poses challenges in devising effective prevention and control measures for ASFV infections (Andrés et al., 2020).
The core shell of the ASFV viral particle is composed of processed products derived from two polyprotein precursors: pp220 and pp62. Between the two, polyprotein pp62 plays a crucial role in the proper assembly and maturation of the viral core. The polyprotein is consist of p15, p35, and p8 with a relative molecular weight of 60.5 kDa (Urbano et al., 2021; Jia et al., 2017), and is encoded by CP503R gene. Its sequence exhibits a high degree of conservation across different ASFV strains (Zhong et al., 2022). Its presence is necessary for proper assembly and maturation of viral particle’s core. In its absence during viral morphogenesis, a homogeneous population of empty particles is produced. Given its vital role in ASFV pathogenesis, eliciting a successful immune response against pp62 can potentially prevent infection and serve as effective immunotherapeutic agent in combating ASF (Suárez, 2010).
The field of immunoinformatics has greatly enhanced the current understanding of diagnosis, pathogenesis, immune response, and computational vaccinology (Oli et al., 2020). With the recent advancements in immuno-informatics tools and databases, the search for protein sequences and the identification of MHC-binding motifs for the epitopes have become more efficient and cost-effective. This study took the advantages that computational vaccinology approach can offer in strategically and efficiently mapping candidate CD8+ epitopes in ASFV. The main objective of the study was to identify highly conserved epitopes derived from the pp62 polyprotein of ASFV that can bind promiscuously to dominant swine leukocyte antigen (SLA). Immunoinformatics tools and databases were utilized to obtain protein sequences and identify epitopes which serve as promising candidates for the development of epitope-based vaccines. It is anticipated that the epitopes identified in this preliminary research will undergo in vitro and in vivo studies to determine their suitability for inclusion in future epitope-based vaccines against ASF.
Materials and methods
Retrieval and identification of highly conserved pp62 sequences
To generate information on the post-translationally modified sites of the pp62 polyprotein, related data on its sequence were obtained from the Universal Protein Resource (UniProt) server (https://www.uniprot.org/). The ASFV protein sequences were collected from the National Center for Biotechnology Information (NCBI) protein database (https://www.ncbi.nlm.nih.gov/protein/) on March 21, 2022, by filtering the sequences based on a length of 530–534 residues. To eliminate redundant protein sequences (Fu et al., 2012), sequences from NCBI were uploaded and analyzed in CD-HIT server (https://sites.google.com/view/cd-hit?pli=1) using 1.00 as identity cut-off value. Unique, representative sequences obtained from the CD-HIT server were subsequently aligned in Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Conserved sequences were identified using the Protein Variability Server (PVS) tool (http://imed.med.ucm.es/PVS/). Consensus sequence was chosen as the representative sequence for further analysis (Garcia-Boronat et al., 2008). Conserved fragments containing at least nine residues were included, while fragments with a Shannon variability scores ≥ 0.1 were masked and excluded. The resulting fragments were tabulated based on their alignment positions in the sequence.
CD8+ epitope mapping
The highly conserved sequences obtained were analyzed in the Immune Epitope Database (IEDB) using the Proteasomal cleavage/TAP transport/MHC class I combined predictor (http://tools.iedb.org/processing/) and the netMHCcons prediction method. The netMHCcons method is a consensus prediction method that combines netMHCpan, PickPocket, and netMHC (ANN) to provide more reliable results (Karosiene et al., 2012). The prediction method included all 45 available SLA alleles to generate nonameric CD8+ epitopes. The immunoproteasome type was used for proteasomal cleavage prediction, and the default parameters were used for TAP transport prediction. The tabulated results from these prediction methods include the proteasome score, TAP score, MHC score, processing score, and MHC-binding score for each peptide. Peptides with an MHC IC50 scores ≤ 500 nM (Fleri et al., 2017; Jensen et al., 2018) and positive TAP and proteasome scores were selected for subsequent analysis.
To assess for potential cross-reactivity, mapped epitopes were compared against available protein sequences of domestic pigs (Sus scrofa domesticus) using BlastP (Protein Basic Local Alignment Search Tool) in NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). The database search excluded model, uncultured, and environmental sample sequences. Epitopes that did not exhibit significant sequence similarities with the sequences in the databases were selected as candidate ASFV pp62 CD8+ epitopes.
In silico validation of candidate epitopes
To further support the binding ability of mapped candidate epitopes with the dominant swine leukocyte antigen (SLA-1*0401), molecular docking analysis were conducted. Currently, it is the only available SLA I PDB crystal structure in all the protein structure databases. The crystal structure of SLA-1*0401 (PDB 3QQ3) was downloaded from the Protein Data Bank. The structure contains an influenza epitope (NSDTVGWSW) bound to the peptide-binding groove of SLA-1*0401; thus, the influenza epitope was removed from the complex to enable docking of each candidate epitope in GalaxyPepDock server (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=PEPDOCK). Other unrelated heterostructures were also eliminated as part of the complex preparation for the docking procedures (Hernández-Santoyo et al., 2013).
Resulting complexes with the highest scores were retrieved and refined using the RefineComplex tool (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE). Refined complexes were visualized using Chimera X version 1.3. To further analyze the favorability and stability of binding, the binding energy and dissociation constant were estimated at 38.6 using the PRODIGY server (https://wenmr.science.uu.nl/prodigy/). These values were then compared to a positive reference peptide (NSDTVGWSW).
To further assess the stability and flexibility of the SLA-epitope complex formation, molecular dynamics (MD) simulation was performed. The simulation was carried out using the OpenMM toolkit on the Google Colab framework. The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) plots were obtained to analyze the system’s behavior over time (Arantes et al., 2021). The AMBER MD setup was utilized for the MD simulation procedures. Solvation parameters were determined using the Amber ff19SB force field while the OPC water model was used in constructing the topology and state file (Tian et al., 2020). The simulation duration was set to 5 ns, and an energy minimization procedure consisting of 1000 steps was performed before the simulation. The system was neutralized with a 0.15 M NaCl concentration. A force constant of 500 kJ/mol was used to maintain a stable system at a temperature of 312.32 K and a pressure of 1 bar.
Results
Highly conserved sequences in ASFV pp62 polyprotein
Computational analysis of pp62 polyprotein sequence aided in the identification of 12 highly conserved fragments with a Shannon variability index ≤ 0.1. These fragments exhibit minimal variability among different strains of ASFV. The length of these highly conserved fragments varies from 11 to 86 residues. Table 1 provides the detailed information for the positions of these highly conserved fragments along the pp62 amino acid sequence.
Table 1
Highly conserved fragments of ASFV pp62 polyprotein
Highly conserved and promiscuous CD8+ epitopes in ASFV pp62
Table 2 presents a total of 21 highly conserved CD8+ epitopes which were identified from the analysis of conserved fragments of pp62. It includes information on the sequence locations of the epitopes, TAP scores, proteasome scores, and IC50 scores, along with their corresponding SLA binders. These epitopes were selected based on MHC IC50 scores and positive TAP and proteasome scores for effective MHC-binding ability.
Table 2
Highly conserved CD8+T-cell epitopes from ASFV polyprotein pp62
Moreover, among the 21 CD8+ epitopes identified, five epitopes (FINSTDFLY, GTDLYQSAM, STDFLYTAI, TEFIKVLPL, and TLESLILPF) were classified as promiscuous because they can also bind to more than one MHC allele (Bhasin and Raghava, 2007).
Evaluation of binding energy and affinity to SLA I
The study focused on the molecular docking of candidate epitopes with SLA-1*0401, including the four promiscuously binding epitopes from pp62: FINSTDFLY, GTDLYQSAM, STDFLYTAI, and TLESLILPF. The positive reference used in the study was the influenza epitope (NSDTVGWSW) which is originally bound to SLA-1*0401. Figure 1 illustrates the 3D structures of the epitope-SLA complexes, showcasing the binding groove of the swine MHC I.
Fig. 1
Positive reference epitope NSDTVGWSW (A) and candidate CD8+ epitopes FINSTDFLY (B), GTDLYQSAM (C), STDFLYTAI (D), and TLESLILPF (E) docked on the peptide binding groove of the swine leukocyte antigen – 1*0401 PDB structure
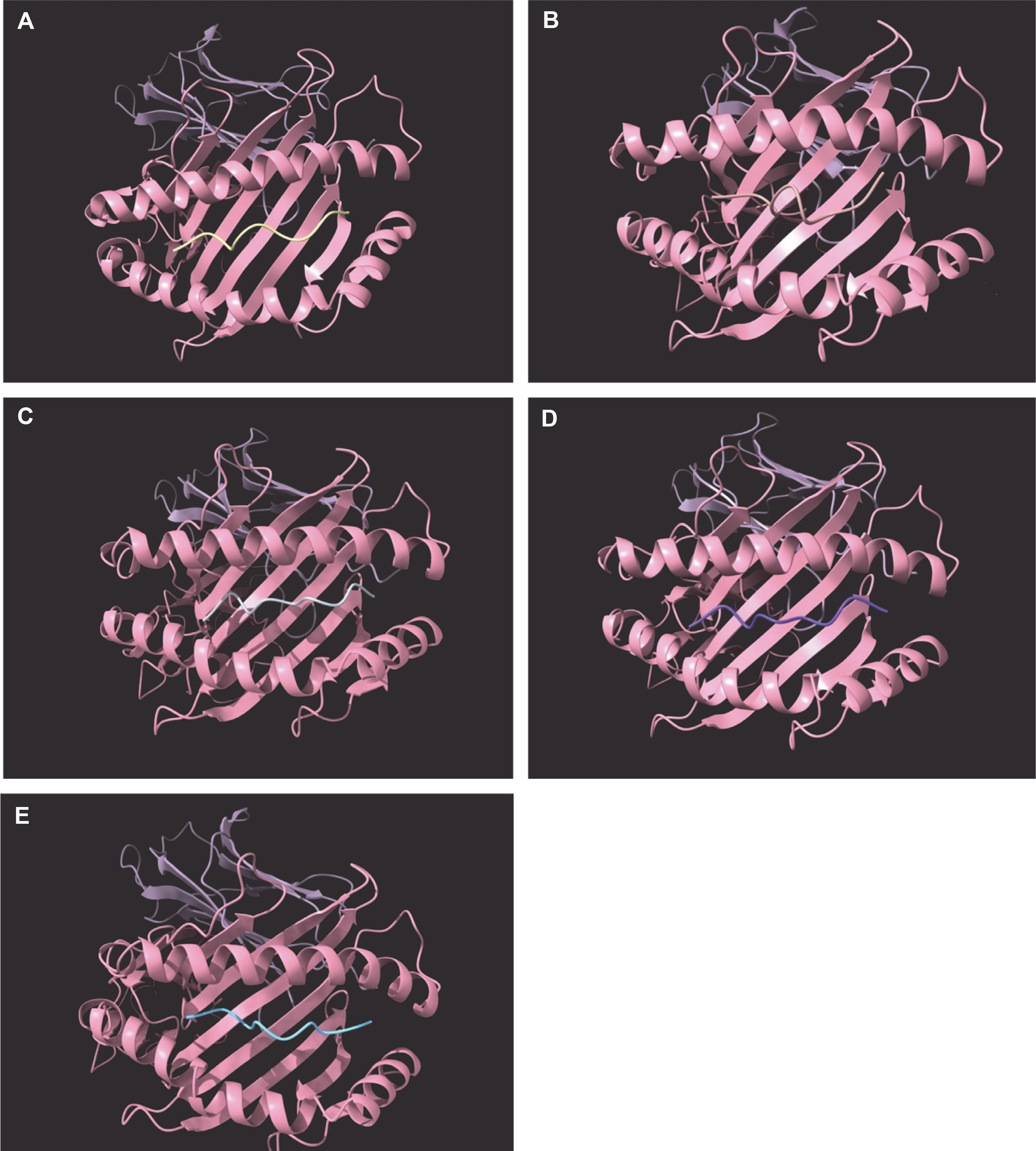
To visualize the interaction between the SLA-1*0401-epitope complexes, protein–ligand interactions were observed. Majority of interactions observed were hydrogen bonding between the candidate epitopes and SLA-1*0401, as H-bonds are known to facilitate binding between the protein and the ligand of interest (Chen et al., 2016). Table 3 provides an overview of the protein–ligand interactions in the SLA-epitope complexes.
To assess favorable binding of the epitopes to SLA-1*0401 binding groove, in comparison to the positive reference, the dissociation constant (Kd) and binding energy (Gbind) of each SLA-epitope complex were estimated. Table 4 presents the calculated values for each SLA-epitope complex. Resulting values imply that the candidate epitopes exhibit comparable binding with the positive reference. Small Kd and Gbind values indicate favorable complex formation of each candidate epitope with SLA-1*0401.
Table 3
Protein–ligand interactions in SLA – epitope complexes
Table 4
Binding energy and binding affinity of SLA 1*0401 – epitope complexes
| Docked epitope | Gbind [kcal/mol] | Kd [mol/l] |
|---|---|---|
| FINSTDFLY | −8.5 | 1.1 × 10−6 |
| GTDLYQSAM | −10.8 | 2.8 × 10−8 |
| STDFLYTAI | −11.5 | 8.1 × 10−9 |
| TLESLILPF | −11.9 | 4.3 × 10−9 |
| NSDTVGWSW | −12.2 | 2.9 × 10−9 |
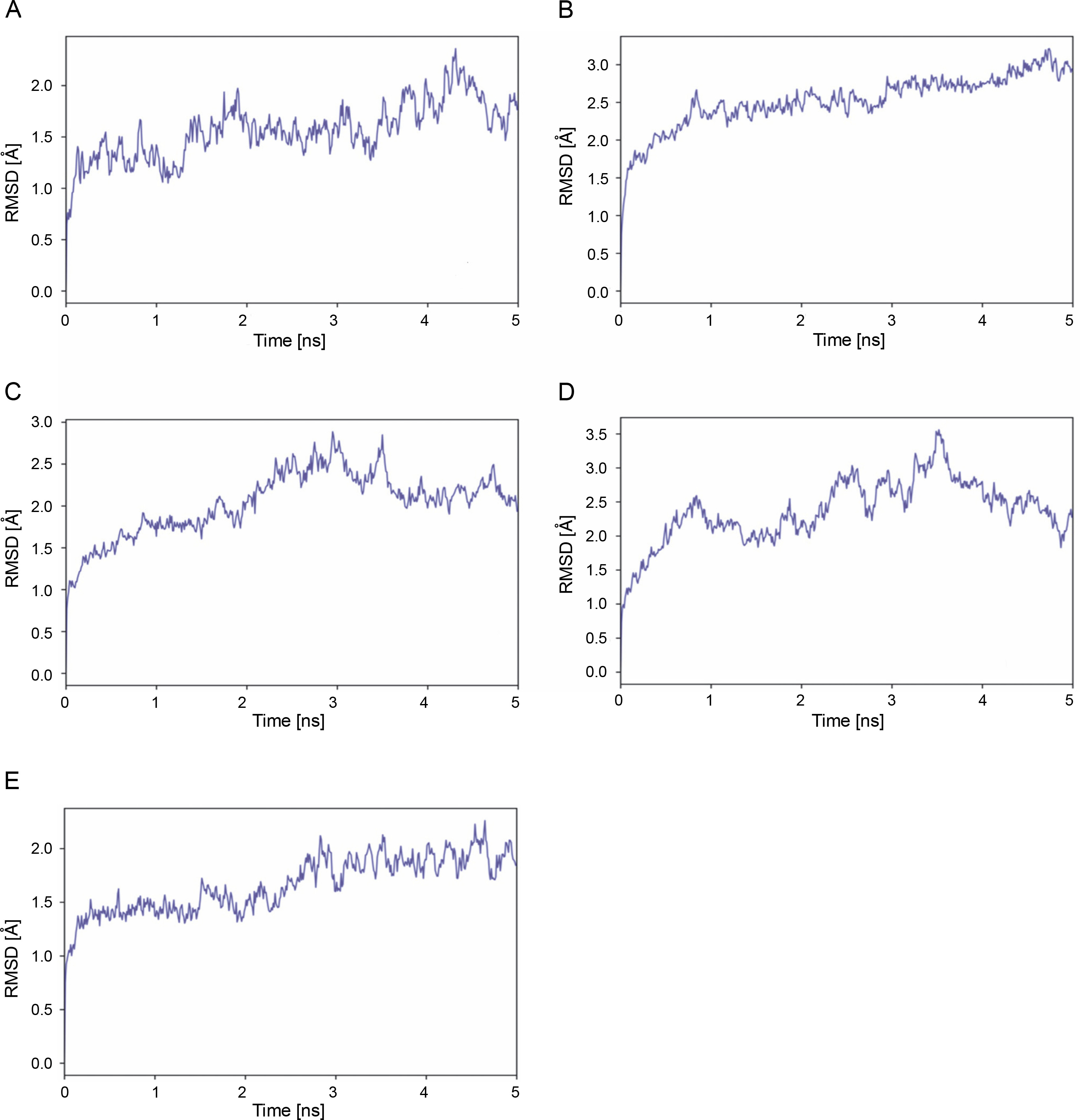
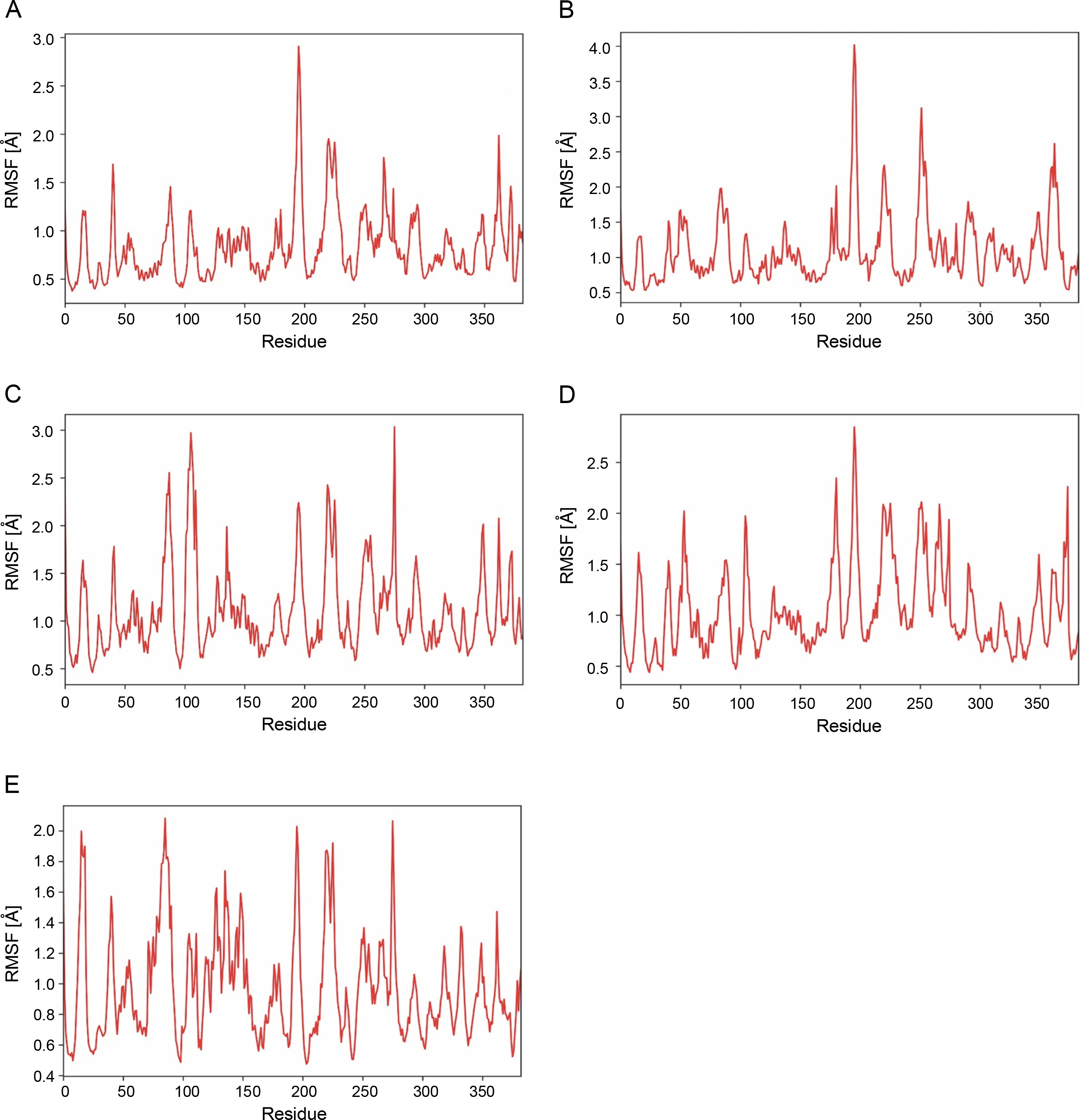
MD simulation was performed to further assess stability and flexibility of epitope-SLA complex formed. The simulation resulted in average RMSD values 1.66, 2.03, 2.37 and 2.52 Å, for peptide-SLA complexes TLESLI LPF, GTDLYQSAM, STDFLYTAI, and FINSTDFLY, respectively (Fig. 2). Moreover, average RMSF values for TLESLI LPF, STDFLYTAI, FINSTDFLY, and GTDLYQ SAM, are 1.88, 2.03, 2.27, and 2.69 Å, respectively (Fig. 3).
Discussion
The lack of commercially available vaccines as prophylactic and therapeutic agents against ASFV has motivated the researchers to identify potent peptides that can be incorporated in future vaccine formulations. Due to known high mutation rates of viral genomes, one of the important strategies to consider in designing vaccines that are capable of inducing long-term immunity is the identification and epitope-mapping of highly conserved sequences. This approach can overcome the challenges associated with epitope immune evasion in rapidly mutating infectious diseases. With this essential step in mind, this study chose to focus on CD8+ T-cell epitopes mapping using the highly conserved sequences of the ASFV pp62 polyprotein.
The sequence conservation analysis employed a Shannon variability threshold ≤ 0.1. Sequences possessing a Shannon entropy of H ≤ 1.0 is considered to be highly conserved (Liu and Bahar, 2012). Findings of the study showed that mapped CD8+ epitopes occupy highly conserved positions in the pp62 polyprotein sequence (Criscuolo and Gribaldo, 2010) which imply that these conserved epitopes can potentially induce long-term adaptive immunity in swine.
Cytotoxic T-cells play crucial roles in providing immunity against ASFV by eliminating virally infected cells. CD8+ T-cells recognize antigenic peptides associated with MHC class I molecules on the surface of virus-infected target cells (Turgeon, 2014). This study successfully identified epitopes that can promiscuously bind to the most common SLA I alleles; thereby, providing broader coverage of the global swine population. MHC polymorphism can affect the interaction between epitopes and the MHC binding groove (Ebrahimi et al., 2019). Thus, the promiscuity of epitopes is another important factor to consider in designing epitope-based vaccines. The binding of these epitopes with the most dominant SLA I allele further supports wider swine population coverage.
A previous study had identified and analyzed highly conserved CD8+ T-cell epitopes of ASFV pp220, pp62, p72, p30, and CD2v. Similarly, researchers in this study docked epitopes that can bind to SLA-1*0401 However, the study was limited to using only the three most common SLAs (SLA-1*0101, SLA-1*0401, and SLA-1*0801) in determining candidate epitopes (Herrera and Bisa, 2021). In contrast, the current study included all SLAs available in the databases to comprehensively map highly conserved CD8+ pp62 epitopes. Moreover, this is the first study to identify and analyze a set of highly conserved CD8+ T-cell epitopes in the ASFV polyprotein pp62 that are also promiscuous binders of 45 various SLA alleles. The researchers identified 21 candidate epitopes with MHC IC50 values ≤ 500 nM which indicate strong binding affinity between the candidate epitope and its corresponding SLA-binder (Fleri et al., 2017; Jensen et al., 2018).
Analysis of candidate epitopes generated other valuable data including proteasome scores which predict the efficiency of C-terminus generation, and the TAP scores which evaluate the ability of the peptide’s N-terminal prolonged precursor. A more positive TAP score indicates high substrate affinity and MHC scores for the candidate epitopes (Abele and Tampe 1999; Fleri et al., 2017).
In vaccine design, safety profile is equally important as vaccine efficacy. To assess the safety profile of mapped CD8+ peptides, all mapped epitopes underwent filtering process in BLASTp against the collection of protein sequences from domestic pigs (Sus scrofa domesticus). This selective step aimed to identify potential cross-reactive epitopes and prevent potential adverse reactions. The BLASTp analysis of the candidate epitopes resulted in E-values ranging from 2.2 to 75. Peptides with E -values < 1.0 × 10−30 are considered crossreactive (Hileman et al., 2002). Using this parameter, the results from this study suggest that all candidate ASFV CD8+ epitopes are less likely to exhibit crossreactivity. This finding is a significant step as it helps mitigate the risk of autoimmune issues that could arise from administering the candidate epitopes in swine.
Furthermore, the analysis of peptide–SLA I interactions revealed that the presence of hydrogen bonds contribute favorably to stability of complex formation (Pace et al., 2014). These hydrogen-bond pairings promote high-affinity receptor–ligand interactions by reducing competitive interference with water molecules (Chen et al., 2016). The strength of these bonds is characterized based on the donor–acceptor distances. Interactions with donor–acceptor distances ranging from 2.2 to 2.5 Å are considered “strong, mostly covalent”. Distance range of 2.5–3.2 Å is categorized as “moderate, mostly electrostatic”, while distance range 3.2–4.0 Å is regarded as “weak, electrostatic” interactions (Jeffrey, 1997).
The estimated dissociation constant values (Kd) for GTDLYQSAM, STDFLYTAI, and TLESLILPF are < 100 nM which indicate favorable epitope binding. Kd values > 100 nM are classified as having larger dissociation rates (Motulsky and Neubig, 2010) where complex dissociation is more likely. Additionally, the binding energies ( Gbind) for all the docked epitopes are negative which further underline the spontaneity of epitope-SLA I complex formation.
The stability and flexibility of docked immune complexes were assessed using RMSD and RMSF parameters. RMSD analysis was used to evaluate deviations during the simulation, with smaller deviations indicating higher stability of the protein complex (Aier et al., 2021). The RMSD value of the complex formation reflects the stability of the complex, as it is influenced by the binding of the ligands (Mawere and Kumar, 2014). Fluctuations within the range 1.0–3.0 Å are considered acceptable and are expected to occur (Shawan et al., 2021). Results indicate that complex formation is stable based on the fluctuation values within a specific period. It is also important to note that the binding of ligands affects the RMSD of protein backbone (Mawere and Kumar, 2014). In terms of structural flexibility, the RMSF plots of the candidate epitopes indicate the fluctuations of the residues within the complex. An RMSF value with fluctuations above 4 Å in the amino acids of the binding site may suggest suboptimal binding, as the residues do not form strong interactions with the small molecule (de Vita et al., 2021). Upon evaluation of RMSD and RMSF values of each epitope–SLA complex analyzed in this study, results showed that the binding of candidate epitopes with their corresponding SLA are within the acceptable fluctuation range which suggest the formation of stable immune complexes. Taken together, estimated values of these parameters can further support the efficacy of identified candidate CD8+ epitopes in potentially inducing cytotoxic immune response against ASFV in swine.
Conclusion
This study successfully identified a set of highly conserved and promiscuous CD8+ epitopes in the ASFV polyprotein pp62 which can potentially induce immunogenicity upon incorporation in future ASF vaccine formulations. All candidate epitopes exhibited acceptable binding affinity and formed flexible yet stable epitope–SLA complexes, emphasizing their potential immunogenicity. More importantly, the low probability of cross-reactivity with the swine proteome highlights the safety profile of candidate peptides. Nevertheless, to establish the efficacy of these candidate epitopes, further evaluations through subsequent in vitro and in vivo studies are recommended.