Introduction
Gnetum is a genus belonging to the group of gymnosperms, and it grows in diverse forms and habitats such as tropical evergreen trees, shrubs, or lianas. Due to the association of the fossils of some Gnetum species with those of extinct scorpionflies, they have been proposed as one of the first groups of plants to be insect-pollinated (Ren et al., 2009). A unique species of Gnetum, namely Gnetum gnemon, resembles the angiosperms in terms of its vegetative structures (Bhat and Binti, 2014). Nutritionally, G. gnemon is very rich in minerals and proteins, which indicates the potential role of this species in providing healthy sustenance in poor provincial regions that have limited access to proteinaceous foods (Bhat and Binti, 2014). It is also known to be rich in antioxidant, tyrosinase inhibitory, antimicrobial, anti-aging, and various bioactive compounds (Barua et al., 2015; Agarwal et al., 2020). The edible parts of G. gnemon include young leaves, inflorescences, and tender tips, which are used as vegetables in Assam, mostly by the Karbi tribe. Seeds are often used as a snack food ingredient by processing them into sundried, flat cakes called emping, which are then fried in oil before consumption (Barua et al., 2015). Among other edible parts, tender leaves are one of the most commonly traded vegetables in ethnic markets in North-East India. Leaves of G. gnemon are cooked with “pholo” (alkaline water) along with dried fish, which is served as a delicacy; the leaves are also eaten fried or cooked with lentil (Terangpi et al., 2013). “Melinjo” (common name for Gnetum ) seeds have high nutritive value and are consumed as a major food item by the Indonesian population as an additional ingredient in soups (viz., “Sayur Asem”) or boiled with spices and vegetables (Narayanan et al., 2015; Ahmad et al., 2018). As a foodstuff, Melinjo seeds have been found to have no toxic effects even when consumed for a long period of time (Tatefuji et al., 2014).
MicroRNAs (miRNAs) are 21-24 nucleotide long, highly conserved, non-coding small RNAs originating from a self-complimentary stem-loop precursor sequence, and they mediate transcriptional or post-transcriptional gene regulation (Nithin et al., 2015). These short RNA sequences also play a pivotal role in manifold facets of plant development and adaptive potentiality of plants to biotic and abiotic stresses (Jones-Rhoades et al., 2006; Vishwakarma and Jadeja, 2013; Bordoloi et al., 2021; Krishnatreya et al., 2021). miRNAs from plants were first recognized in early 2002 in Arabidopsis thaliana (Reinhart et al., 2002), following which thousands of plant miRNAs were identified in different plants and submitted to databases such as the miRBase Sequence database (http://www.mirbase.org) (Griffiths-Jones et al., 2006).
The identification of miRNAs and their target genes from plants has been widely investigated in the last few years (Pirro et al., 2019; Xu et al., 2020; Panda et al., 2014). To identify miRNAs and their target genes and to perform functional analysis, various strategies are usually used, such as genetic screening (Xun et al., 2019), construction of a small RNA library, or computational approach-based identification from ESTs (Krishnatreya et al., 2020). The computational biology-based approach serves as a useful asset to identify conserved miRNAs in organisms for which complete genome sequences are not yet available. This approach is also used to study the conservation and evolution of such miRNAs among different species (Vishwakarma et al., 2013). Identification of miRNAs from the published ESTs has been shown to be effective for revealing new miRNAs from plants that are socio-economically important; lacking significant genomic resources (Lu et al., 2010). Most of the mature miRNAs in plants are evolutionarily conserved from species to species within the plant kingdom (You et al., 2017). This conserved nature of plant miRNAs has greatly enhanced the identification of conserved miRNAs using EST sequences. There are, however, no reports of identification of miRNAs from G. gnemon till date. In the present study, we identified conserved miRNAs in G. gnemon ESTs available from the NCBI database. These identified miRNAs of G. gnemon may even arbitrate the cross-kingdom miRNA-mediated human gene target networks that are potentially regulated through the target gene cleavage, based on the significant target specificity between the identified miRNAs of G. gnemon and human gene transcripts.
Materials and methods
Reference set of miRNAs and sequence database
To identify the potential of conserved miRNAs, the sequences of 38,589 plant miRNAs previously identified from different plant species were downloaded from the miRBase database (http://www.mirbase.org, release 22.1, March 2018). These sequences were used as query sequences to perform BLAST analysis against the G. gnemon ESTs. Publicly available 10,725 ESTs (as of August 1, 2019) of G. gnemon were downloaded from the NCBI (https://www.ncbi.nlm.nih.gov/). A local database for standalone BLAST was constructed for the G. gnemon ESTs by using a locally installed NCBI-Blast+ v-2.2.26 command line application (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/) (Altschul et al., 1997). Non-redundant (NR) protein sequences were used from the NR protein database of NCBI (ftp://ftp.ncbi.nlm.nih.gov/blast/db/).
Identification of putative miRNAs
Sequence identification of precursor miRNAs was performed using a comparative sequence-based homologue search and a secondary structure analysis. Mature miRNA sequences were used for BLASTn search as they are evolutionarily more conserved than their precursor sequences. miRNA sequences were used as queries and subjected to NCBI-Blast+ against the G. gnemon EST database. A maximum of two allowed mismatches of threshold e -value of < 0.001 and word-size value of 7 were set for Blast+ analysis. After removing redundant sequences, the ESTs with matched hits were subjected to a Blastx analysis with the NR protein database, and the non-protein coding sequences were retained for further analysis of RNA secondary structure using the Zuker folding algorithm by Mfold v3.5 (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form) (Zuker, 2003). The following parameters were used in defining the sequences as miRNA homologs: 1) the sequence should fold into an appropriate stem-loop secondary structure, 2) the miRNA should exist in one arm of the hairpin structure, 3) the mature miRNA and its complementary miRNA* sequence should not have more than 5 mismatches, and 4) the value of the Minimal Folding free Energy Index (MFEI) of precursor miRNA structures should not be less than 0.5 and should have a high Minimal Folding free Energy (MFE) value. MFE is the negative equivalent of the ΔG value (Zhang et al., 2006). The MFEI value was calculated using the following formula proposed by Zhang et al. (2006):
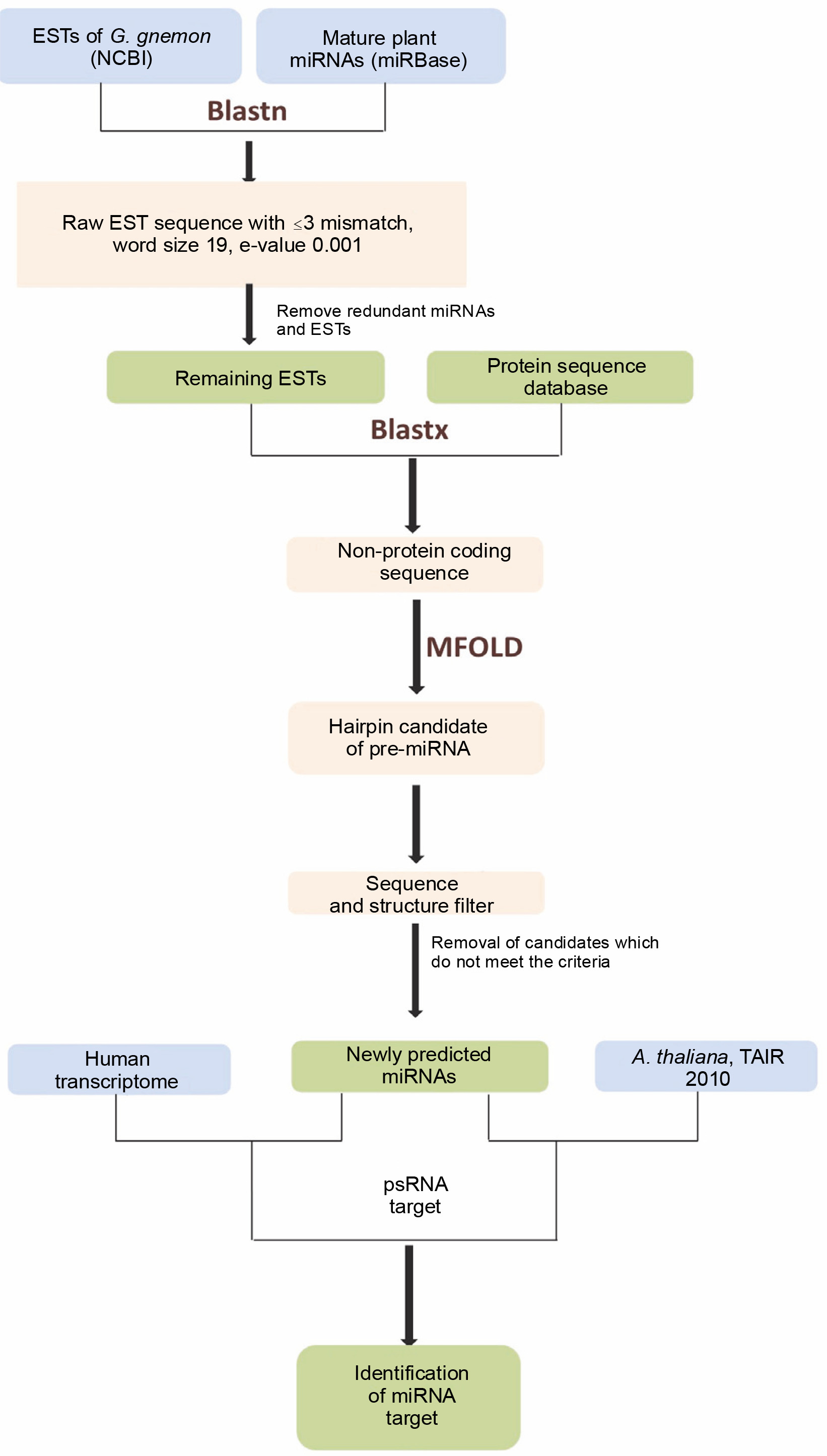
* Adjusted Minimal Folding free Energy
Prediction of putative target genes
The perfector near-perfect complementarity of miRNAs and their target mRNAs in plants has greatly simplified the identification of miRNA targets. This strategy is applied to search the targets of identified miRNAs by a homology algorithm. In the present study, A. thaliana transcript, TAIR version 10 available at psRNATarget (http://plantgrn.noble.org/psRNATarget/) was used as a reference for finding the targets of the candidate miRNAs as no genomic references of Gnetum species are available in public domain. To examine the possible cross-kingdom interaction exerted by miRNAs of G. gnemon on human genes, a target analysis was performed against the human transcriptome available at psRNATarget. A plant small RNA Target Analysis Server, namely psRNATarget, was used for predicting the targets of the newly identified miRNAs by using Schema V2 (2017 Release) with the maximum expectation value threshold as 3 and the remaining values set as default (Dai et al., 2018). A maximum of two mismatches were allowed in the complementary region of target genes with the miRNAs, whereas the mismatch inhibition was maintained at the 10th and 11th nucleotide positions along the aligned region. Figure 1 shows the pipeline followed for miRNA identification and target gene prediction. Information regarding the disease association and the function of human target genes was collected from GeneCards – a human gene database (https://www.genecards.org/) and MalaCards – a human disease database (https://www.malacards.org/).
Gene Ontology, KEGG pathway, and phylogenetic analysis
Annotations of the target genes for both plant and cross-kingdom human targets were carried out using a Blastx analysis with a threshold e -value of 10–3 against the NCBI NR protein database. Blast2go version 5.2 (https://www.blast2go.com/blast2go-pro/) was used for the gene ontology and the KEGG pathway analysis of the annotated target genes in order to assess the traits that may be affected by the expression of the identified miRNAs (Conesa and Götz, 2008). The phylogenetic tree generated from the identified miRNA precursors was constructed using MEGA7. The precursor sequences of the family members of the miRNAs belonging to other plant species were downloaded from the miRBase and collated with the G. gnemon miRNA precursors. A multiple sequence alignment was performed using the MUSCLE algorithm, and the phylogenetic tree was generated using the Maximum likelihood method.
Results
miRNA identification and characterization
From a total of 10,725 published ESTs of G. gnemon, 144 ESTs showed homology with the previously deposited sequences of miRNAs in miRBase 22.1, belonging to various miRNA families. These sequences were obtained after following the criteria of Axtell and Meyers (2018) for plant miRNA annotation, wherein only those miRNAs of ≥19 nucleotides in length are filtered and retained. Further removal of redundancies in miRNAs and the ESTs yielded 142 potential miRNA sequences. A Blastx analysis of these ESTs against the NCBI NR database led to the identification of 56 sequences as non-coding sequences, which did not show homology with any protein coding gene. These 56 sequences were retained for further analysis of the secondary structure of their precursors.
miRNA secondary structure
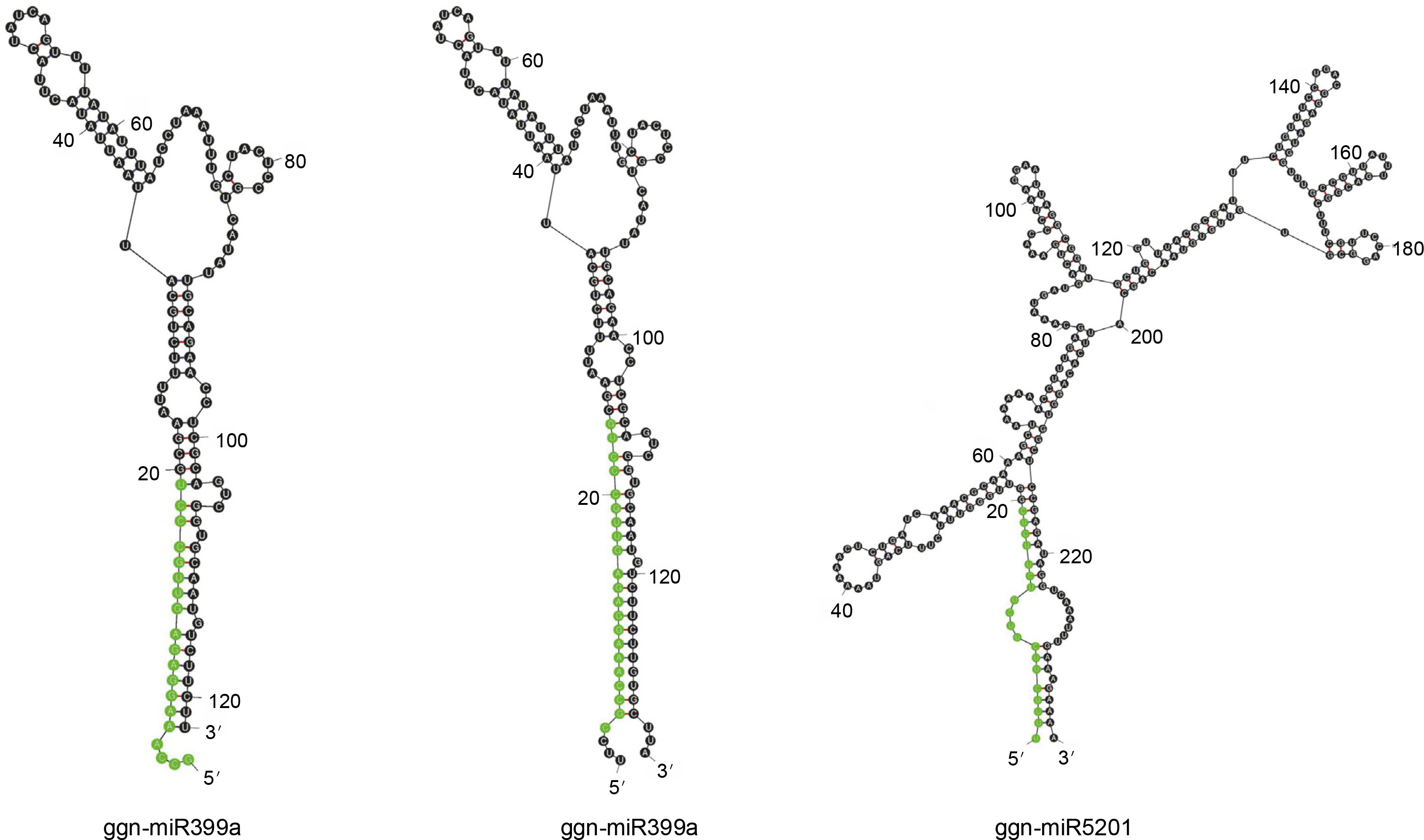
The 56 potential non-coding sequences were subjected to a structural validation analysis using Mfold v3.5 to predict the miRNA secondary structure. The miRNAs meeting the following criteria were considered valid for further analysis of their targets: 1) a valid stem-loop hairpin precursor, 2) the presence of complementary miRNA* sequence in the precursor with less than 6 mismatches, and 3) the MFEI value greater than 0.5. Twenty such conserved miRNAs were identified as belonging to two miRNA families. Nineteen members of miR399 were identified from various plant species, while miR5021 had only a single member from A. thaliana. On the basis of the stem-loop precursors, the identified miRNAs for G. gnemon were classified to be represented by two members of ggn-miR399 (ggn-miR399a and ggnmiR399b) and one member of ggn-miR5021 (Fig. 2). The ΔG values ranged from –58.05 to –24.52 kcal/mol (Table 1). It is often considered that the lower the value of ΔG, the higher is the thermodynamic stability of the miRNA precursor (Bonnet et al., 2004). A lower value of ΔG corresponds to higher MFE and MFEI values as MFE is equivalent to –ΔG (Zhang et al., 2006). The miRNA characterization indicates that the precursor length of miRNAs varied from 121 to 240 bases and the mature miRNA lengths ranged from 19 to 21 nucleotides (Table 1).
Table 1
Identified putative miRNAs of G. gnemon from ESTs
| Accession no | miRNAs | Mature miRNA sequence | PL* | ML** | (C + G)% | MFE | AMFE | MFEI |
|---|---|---|---|---|---|---|---|---|
| CB081553.1 | sly-miR399 | GCCAAAGGAGAGUUGCCCU | 122 | 19 | 38.5 | 24.52 | 20.09 | 0.52 |
| tcc-miR399a | CGCCAAAGGAGAGUUGCCCUG | 133 | 21 | 39.1 | 27.62 | 20.76 | 0.53 | |
| ath-miR399b | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| csi-miR399b-3p | GCCAAAGGAGAGUUGCCCU | 122 | 19 | 38.5 | 24.52 | 20.09 | 0.52 | |
| pab-miR399c | CGCCAAAGGAGAGUUGCCCUG | 133 | 21 | 39.1 | 27.62 | 20.76 | 0.53 | |
| aly-miR399c-3p | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| mdm-miR399d | GCCAAAGGAGAGUUGCCCU | 122 | 19 | 38.5 | 24.52 | 20.09 | 0.52 | |
| csi-miR399d-3p | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| cme-miR399e | GCCAAAGGAGAGUUGCCCU | 122 | 19 | 38.5 | 24.52 | 20.09 | 0.52 | |
| zma-miR399e-3p | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| mes-miR399f | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 38.5 | 27.62 | 20.76 | 0.53 | |
| gma-miR399h | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| vvi-miR399i | CGCCAAAGGAGAGUUGCCCUG | 133 | 21 | 39.1 | 27.62 | 20.76 | 0.53 | |
| zma-miR399i-3p | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| osa-miR399j | GCCAAAGGAGAGUUGCCCU | 122 | 19 | 38.5 | 24.52 | 20.09 | 0.52 | |
| zma-miR399j-3p | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| mdm-miR399k | GCCAAAGGAGAGUUGCCCU | 122 | 19 | 38.5 | 24.52 | 20.09 | 0.52 | |
| mtr-miR399l | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| mtr-miR399p | GCCAAAGGAGAGUUGCCCUG | 133 | 20 | 39.1 | 27.62 | 20.76 | 0.53 | |
| EX943654.1 | ath-miR5021 | UUUUCUUCUUCUUCUUCUC | 240 | 19 | 41.6 | 58.05 | 24.18 | 0.58 |
Phylogenetic analysis of precursor sequences
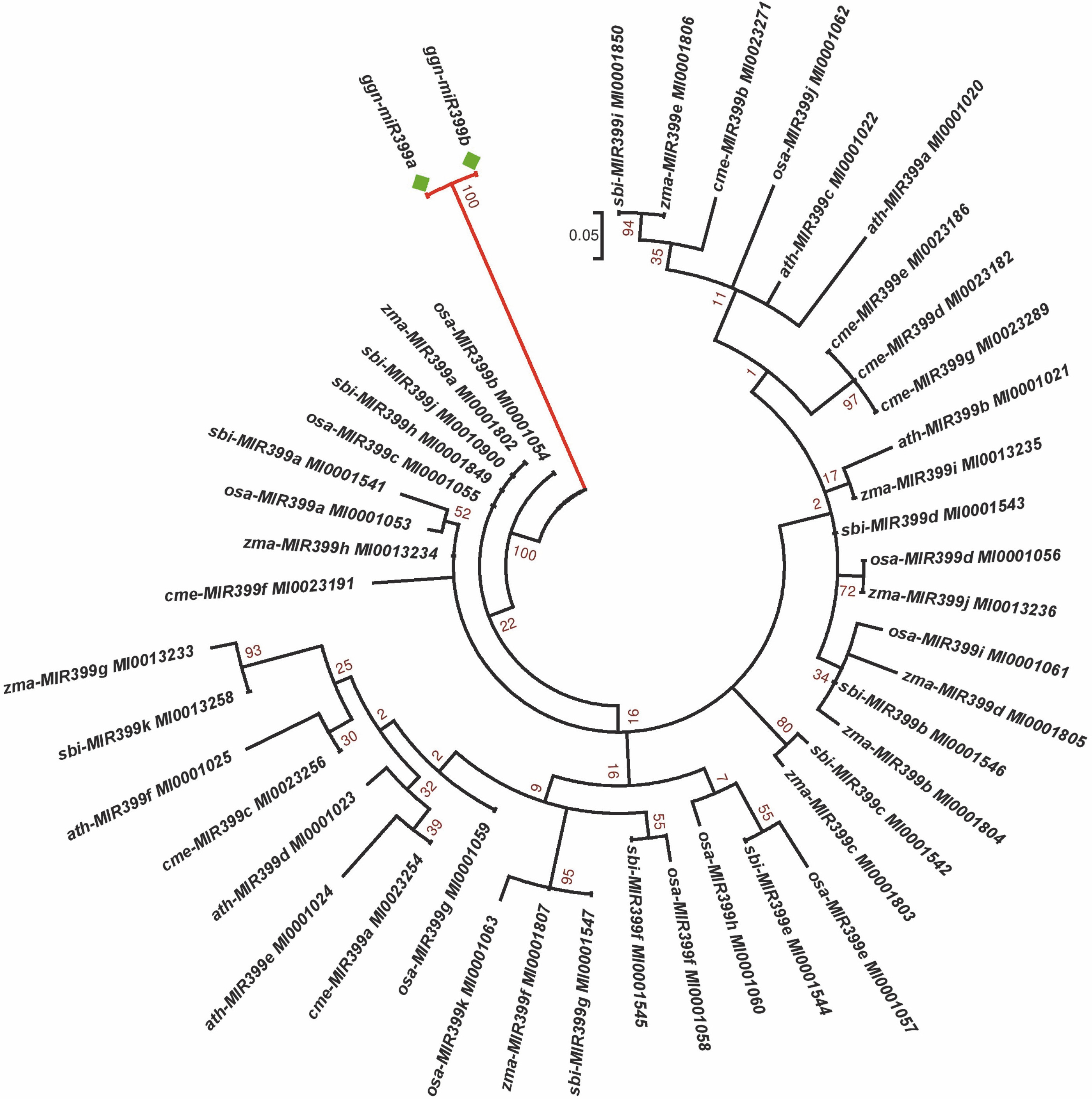
A phylogenetic analysis was performed to understand the relationship of the identified miRNAs in G. gnemon with other plant species available in the miRNA database in order to identify sequences belonging to the same miRNA families (Fig. 3). No miRNAs for G. gnemon have been reported earlier in any public domain. Hence, a phylogenetic tree generated using miRNA precursors will be helpful to understand the evolutionary and conserved nature of the identified miRNAs. Members of the miR399 family have been identified in many plant species, and precursor sequences of miR399 were retrieved from miRBase for A. thaliana, Oryza sativa, Zea mays, Sorghum bicolor, and Cucumis melo (Wang et al., 2020; Gonzalez-Ibeas et al., 2011; Zhang et al., 2011). Since the pre-miR5021 has been deposited only for A. thaliana in miRBase, the phylogenetic analysis was performed only for members of the miR399 family by using the maximum likelihood method. The precursor sequences of the identified miRNA families of G. gnemon formed a distinct cluster from other plant species (Fig. 3).
Prediction and annotation of target genes
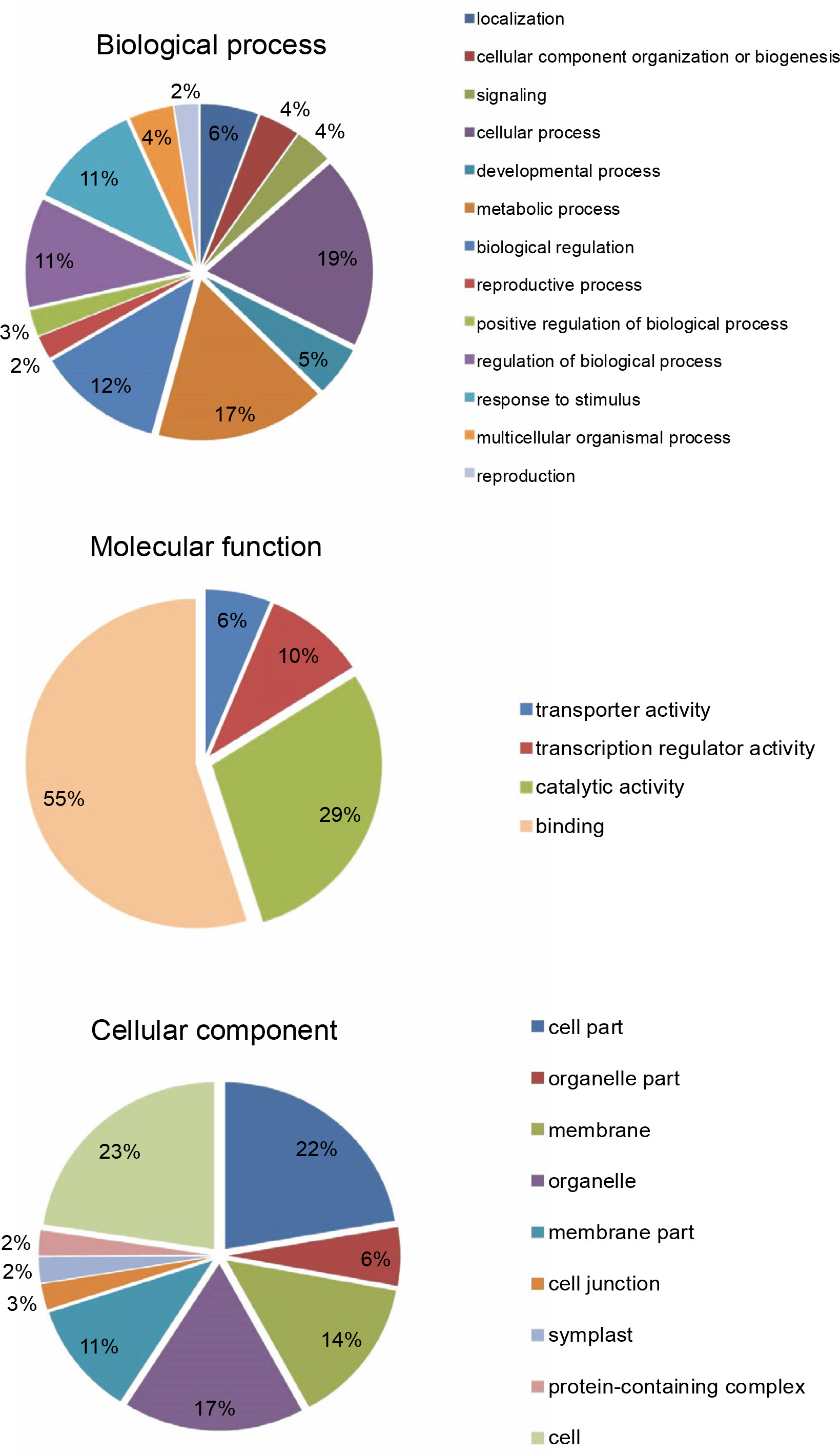
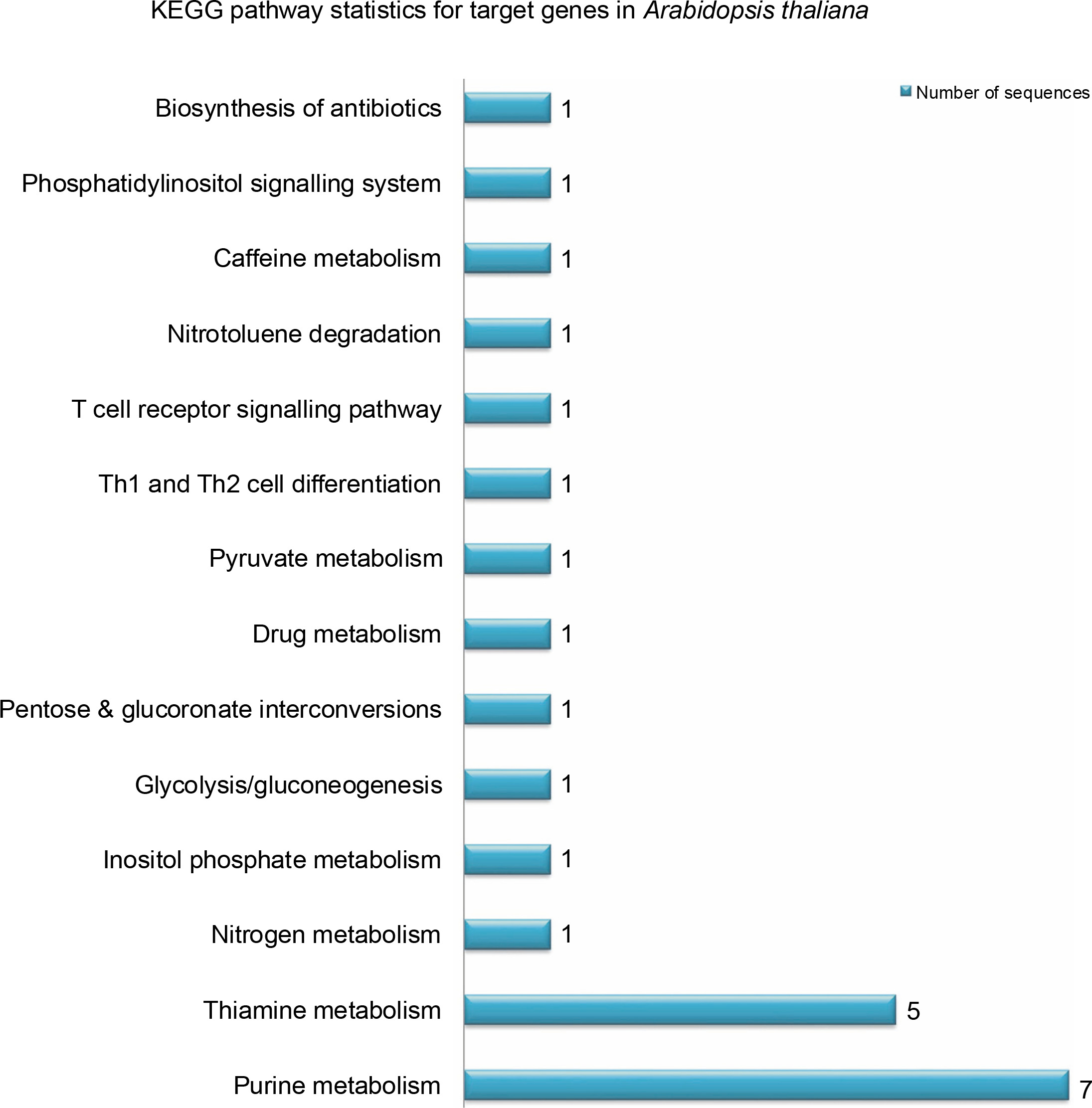
miRNAs regulate the expression of specific genes by binding to mRNA transcripts based on perfect or near perfect complementarity to promote RNA degradation, inhibit translation or both (O’Brein et al., 2018). To identify genes plausibly recognized by the potential miRNAs, psRNATarget was used for searching target genes against the A. thaliana transcriptome acquired from TAIR10 and using the default parameters specified in SchemaV2 (2017). After removal of genes with unknown functions, a total of 100 genes (listed in Table 2) were identified as target genes of 20 identified miRNAs. To further realize the regulatory potential of the identified miRNAs, the target genes were subjected to the Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis by using Blast2GO v5.2. The identified target genes were found to participate in 13 distinct biological processes and 4 molecular functions (Fig. 4). Among the predicted targets, 10% genes were sequence-specific transcription factors (TFs), 29% genes were involved in various catalytic functions, and 55% genes acted as sequence-specific or metal-ion binding factors. The two main categories represented among the biological processes were cellular processes and metabolic processes (19% and 17% genes, respectively). A pathway analysis of the target genes using the KEGG online database demonstrated the participation of the identified miRNAs in 14 vital metabolic networks such as purine metabolism, thiamine metabolism, nitrogen metabolism, glycolysis, and caffeine metabolism (Fig. 5). GO reports for each target gene can be accessed from Supplementary Table S1.
Table 2
Predicted target genes of G. gnemon miRNAs against Arabidopsis thaliana
Cross-kingdom target gene prediction
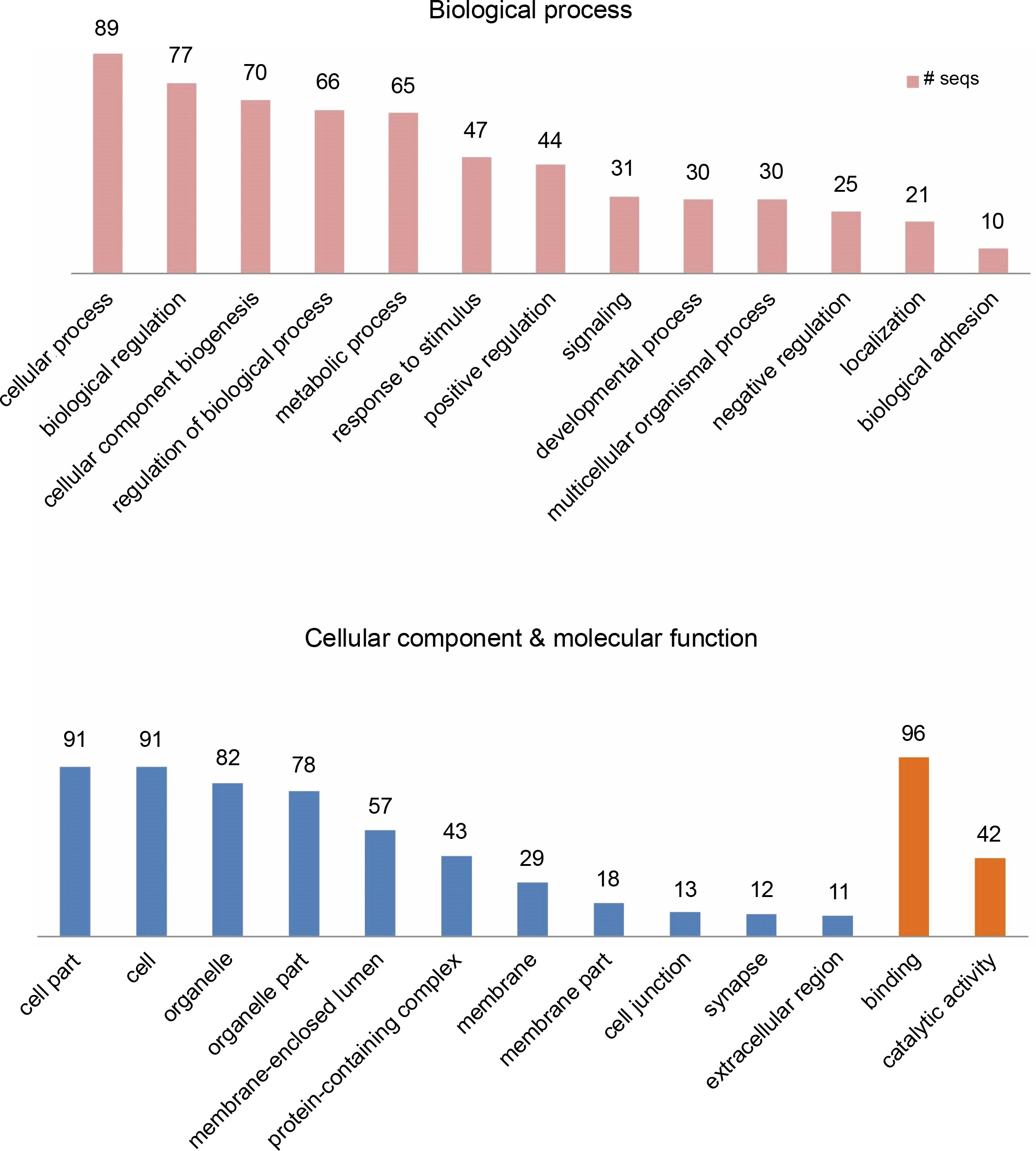
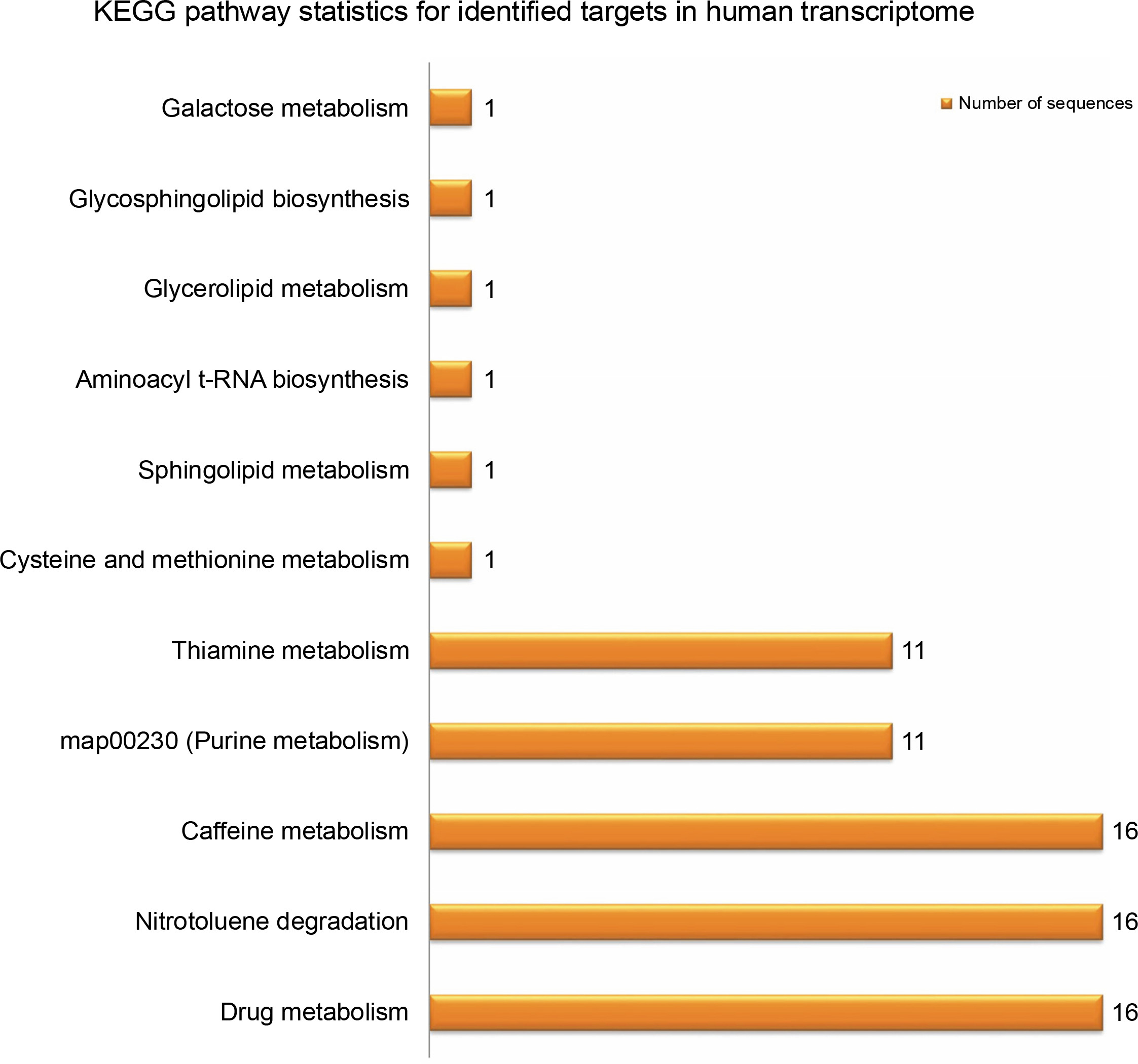
Provided that the human diet is primarily dependent on plants, researchers have already proposed significant potential effects of plant small RNAs after food consumption on human gene expression (Vaucheret and Chupeau, 2011; Li et al., 2018; Sanchita et al., 2018). For better understanding of the potential biological functions of the newly identified G. gnemon miRNAs in human transcriptome, putative target genes were searched using the psRNATarget with default parameters against the Homo sapiens transcripts, Human Genome Sequencing project. A total of 49 probable targets were identified for the 20 identified G. gnemon miRNAs after eliminating genes with unknown or hypothetical functions. All the listed targets exhibited mRNA cleavage mode of regulation of gene expression by the predicted miRNAs (Supplementary Table S2). GO analysis results suggested that G. gnemon miRNAs could be involved in the regulation of 13 broadly defined biological processes (such as regulation of biological processes, cell signaling, and cellular processes) and 2 basic molecular functions (catalytic activity and binding) in humans. The target genes were also found to be part of 11 different types of cellular components (such as cell organelles, cell membrane, and synapse) (Fig. 6). The KEGG pathway analysis of the target genes demonstrated the participation of the identified miRNAs in 11 different biological networks (Fig. 7). These networks are involved in various important pathways such as drug metabolism, caffeine metabolism, and nitrotoluene degradation.
Discussion
Screening and annotation of small RNAs help to interpret the critical roles played by such genetic modulators in the regulation of gene expression and their associated biological processes. Before this study, no comprehensive work was performed on the identification of putative miRNAs from ESTs of any species of Gnetum. This genus is often regarded as a very important plant group due to its significant taxonomic position as a connecting link between two diverse plant forms, namely gymnosperms and angiosperms (Bhat and Binti, 2014). In the present study, we considered all the important criteria such as the MFEI values, mismatch inhibition, and sequence length, which have been used for miRNA identification in other plant species. The MFEI values of the 20 identified miRNAs in our work were just above 0.5, which is in accordance with that reported for some miRNAs identified in other plant ESTs (Wang et al., 2012; Gupta et al., 2015; Patel et al., 2019; Zakeel et al., 2019). The G + C% of the miR399 members ranged from 37.5% to 38.8%, whereas it was found to be more than 41% for miR5021 mature miRNA sequence.
The TFs of plants have been found to be one of the notable targets of G. gnemon miRNAs. ggn-miR5021 and ggn-miR399 have shown a possible cleavage of a range of TFs such as SPL (SQUAMOSA Promoter-Binding Protein-Like) family of TFs, MYB (myeloblastosis) domain protein, and GRAS (named after the first three members: Gibberellic-acid Insensitive (GAI), Repressor of GAI, and SCARECROW). TFs are regarded as master regulators of gene expression in eukaryotes and are responsible for promoting growth and development in plants (Kaufmann and Airoldi, 2018). SPL genes are known to encode plant-specific TFs that play important roles in phase transition, flower and fruit development, plant architecture, gibberellin signaling, sporogenesis, and response to copper and fungal toxins (Stief et al., 2014). SPL gene association with the miRNAs miR156 and miR172 has been found to play a critical role during the juvenile to adult leaf development in A. thaliana (Chen et al., 2010). Members of the MYB DNA-binding domain superfamily protein are involved in many important biochemical and physiological processes in plants (Du et al., 2009). The family of plant-specific MYB genes has immensely contributed to the evolution of physiological or developmental processes specific to plants, especially those involved in responses to fluctuating biotic or abiotic environments (Ambawat et al., 2013). Studies in several plants have revealed that GRAS family TFs serve diverse functions and are involved in various physiological processes. For instance, GRAS family members have been found to regulate GA signaling and the root and shoot radial pattern formation in Arabidopsis (Lu et al., 2020). The formation of nodules is regulated by two GRAS proteins in Medicago trunculata – a leguminous plant (Wang et al., 2018). Our results also show that ggn-miR5021 may also target several genes that encode various F-box proteins. These proteins are known as components of the SCF ubiquitinligase complexes (Skp I, Cullin, and an F-box protein), in which they bind to substrates for ubiquitin-mediated proteolysis (Kipreos and Pagano, 2000). Protein ubiquitination is considered to be a very critical post-translational modification process that is used by eukaryotes for the regulation of various types of cellular processes (Dye and Schulman, 2007). The target gene prediction analysis also reveals another intriguing possibility that ggn-miR5021 and ggn-miR399 may also target three important defense-related genes, namely Leucine-rich repeat receptor kinases (LRR-RKs), TIR-NBS-LRR protein family, and Time for Coffee (TIC) – a circadian clock regulator involved in time-of-day-related regulation of jasmonate signaling (Shin et al., 2012).
miRNAs have also been shown to have beneficial effects on human health. In fact, cross-kingdom delivery of plant miRNAs into the human alimentary canal can be considered as a mode of herbal therapy against many diseases (Sanchita et al., 2018). G. gnemon is also a popular medicinal plant known for its therapeutic properties in treating human ailments, and it is consumed as food among various tribes (Barua et al., 2015; Agarwal et al., 2020). To analyze the potential effectiveness of the exogenous application of miRNAs derived from this edible plant in humans, we analyzed the target specificity of the identified miRNAs against human genes. Among the identified cross-kingdom targets in human transcriptome, 70% genes were found to be DNA-binding, metal ion-binding, or protein-binding factors, and 30% genes possessed various catalytic functions. In the GO analysis, the two main categories represented among the biological processes are biological pathway regulation and biogenesis of cellular components (13% and 11% genes, respectively). Only one TF – a bHLH TF, namely ASCL1 – was found to be a probable target of miR5021. The “achaete-scute complex-like” (ASCL) family or “achaete scute family basic helix-loop-helix transcription factor” is critical for the proper development of the nervous system in humans, and its dysregulation is proposed to play a key role in psychiatric and neurological disorders (Wang et al., 2017). G. gnemon is used as an edible plant in some North-Eastern parts of India and in some other countries including Indonesia and Thailand. It is often considered as an alternative for protein-rich foods because of the abundance of essential amino acids (Ota et al., 2013). Gnetum seeds are known to be rich in stilbenoids – a class of phenolic compounds – including trans-resveratrol, gnetins, and gnemonosides (Kato et al., 2009). Human studies have revealed that trans-resveratrol is beneficial in the management of diabetes and cardiovascular diseases (Brasnyó et al., 2011, Wong et al., 2011). Local tribes attribute many other medicinal properties to the extracts of seeds and leaves of G. gnemon. Evidence confirming the inhibition of viral infection and other diseases in humans, such as cancer and rheumatoid arthritis, has been reported for exogenous plant-based miRNA application (Sanchita et al., 2018). To ascertain any possible medicinal roles that can be attributed to the miRNAs identified in G. gnemon, we assessed the possible target genes that may be related to any human disorders. Table 3 lists the potential target genes of G. gnemon miRNAs predicted in human transcriptome that are associated with some disorders or ailments in humans and their basic functions. Several such genes were identified amongst the putative targets of ggn-5021 and ggn-399, such as Retinitis pigmentosa GTPase regulator (RPGR), LRP2, BCL2, KLF6, Muscle RAS oncogene homolog, and Otoferlin (OTOF) (Table 3). These genes are associated with various human disorders such as different forms of cancer, deafness, and various disorders of genetic and non-genetic origin (Safran et al., 2010; Rappaport et al., 2013). A noticeable feature found in the human target analysis is that some long non-coding RNAs can also act as potential targets of the identified miRNAs, such as EPHA1-AS1 and C1RL1-AS1 (Table 3). This type of activity suggests that a cleavage-mediated degradation of lncRNAs as target gene mimics might help to enhance the expression of the respective genes (Karlik et al., 2019). Studies related to miRNA-lncRNA-mRNA association modules in various organisms have been conducted as a recent purview in the field of biological research that needs a significant amount of evaluation and substantiation (Zhang et al., 2019). The antitumor activity of “melinjo” seeds has also been studied in human tumor models by Narayanan et al. (2015). The potential application miRNA-based drug delivery has already been studied in tumors, cancers, and osteoporotic fractures (Sun et al., 2019; Anthiya et al., 2018; Si et al., 2019). The identification of cross-kingdom target genes provides a future prospect and direction for exploring plausible medicinal characteristics that can be imparted by plant miRNAs to humans.
Table 3
Disease-associated target genes of G. gnemon miRNAs in the human transcriptome
Conclusions
The ESTs of G. gnemon were used to identify miRNAs and to predict their target genes in A. thaliana and humans by using a computational approach. Twenty putative miRNAs, representing two miRNA families, were predicted from the ESTs of G. gnemon. A total of 100 target genes were predicted, representing various TFs, catalytic enzymes, and cofactors in A. thaliana. A cross-kingdom target analysis in the human transcriptome revealed various genes involved in downstream signaling pathways in diverse functional processes and different organs. Moreover, enlisting the probable target genes of Gnetum miRNAs in the human transcriptome will help to provide a basis for investigating the regulatory and medicinal roles that may be played by the miRNAs of this edible plant consumed as food in significant quantities in some tribal areas. Conclusively, these findings may further enhance our understanding of how the plant miRNAs regulate various metabolic and catalytic processes and help to recognize the therapeutic and nutritional potential of the plant. However, the availability of transcriptome sequences of G. gnemon, their miRNA expression analysis, and validation against the transcriptome sequences will be substantial benchmarks to confirm such possibilities.