










Introduction
Abiotic stress severely inhibits crop development and productivity worldwide, and hence, there is a great interest in understanding the various mechanisms by which plants cope with abiotic stress. To overcome abiotic stresses, plants have evolved adaptive developmental and physiological strategies through the regulation of transcriptional networks (Lee et al., 2017). Transcription factors (TFs) are one of the critical components of these networks. TFs perform their regulatory functions by binding to their specific binding sites of 5–25 bp length (Rani et al., 2007; Zhang et al., 2019). Extensive progress has been made in determining regulatory genes involved in stress responses. This could enable to develop plants tolerant to abiotic stress (Galiba et al., 2009).
Cis-acting regulatory elements are critical molecular switches that play a role in the transcriptional regulation of different biological processes (e.g., abiotic stress responses, hormone responses, and development process). It is possible to obtain an accurate knowledge of the regulatory systems in the stress-responsive genes by analyzing the cis-acting elements (Yamaguchi-Shinozaki and Shinozaki, 2005).
Understanding the intricate mechanisms that govern gene expression regulation is critical and challenging (Das et al., 2019). Determining the factors that regulate genes and identifying their related binding sites is the most common approach to achieve it (Wang et al., 2018). Cohen and Leach (2019) implemented in silico analysis of publicly available rice transcriptome data to identify genes and pathways regulated by abiotic stress, biotic stress, and both stress types.
Barley, as one of the oldest cereal crops, is an important food source for livestock as well as a source of food and drink for humans (Newton et al., 2011). It possesses natural tolerance to the most important abiotic stresses such as drought and salinity, which makes it an excellent model in research on abiotic stress (Munns et al., 2006; Sallam et al., 2019). Despite the genetic complexity of abiotic stress tolerance, there exist valuable genes in the genome of barley with potential for useing in biotechnological improvement programs for abiotic stresses. A genome-wide association study (GWAS) of salinity tolerance performed by Mwando et al. (2020) led to the detection of 19 loci consisting of 52 significant salt tolerant-associated markers. In another study, GWAS was performed in spring barley, and 12 genes that regulated traits under drought conditions were recognized (Thabet et al., 2020). Therefore, genomic and transcriptomic data on environmental stress responses of barley can provide valuable information on single and combined abiotic stress tolerance.
Several miRNAs have been confirmed to play a role in stress responses in many plants (Chuck et al., 2009). For instance, next-generation sequencing (NGS) profiling and northern blot analysis confirmed the upregulation and involvement of miR5655 and miR2933b in Arabidopsis under drought stress (Barciszewska-Pacak et al., 2015). The downregulation of mir419 and the upregulation of its target gene confirmed the regulatory role of this miRNA gene in barley (Deng et al., 2015). Transgenic rice lines in which OsmiR156 was overexpressed showed improved cell viability and growth under cold stress (Zhou and Tang, 2019). Moreover, Zare et al. (2019) reported the upregulation of miR414 and miR2102 in two barley genotypes under drought. However, no association was found between these two miRNAs and the two highest upregulated differential expressed genes HvsnLTP (non-specific lipid transport protein) and HvPiP1;4 (N-butyl-N-methylpiperidinium). Accordingly, the investigation of abiotic stress-associated miRNA families in barley provides substantial opportunities to enhance the tolerance of barley to abiotic stresses.
Most of the studies conducted thus far focus only on one type of stress. However, there is a network of pathways regulated by abiotic stresses (Zhu et al., 2013). Therefore, meta-analysis is a useful tool to identify differentially expressed genes (DEGs). An integrative meta-analysis of stress response studies using micro-array gene expression data and a system biology analysis would provide a potential approach for probing the regulatory gene sets. Gene sets such as TFs, transcription regulators (TRs), and protein kinases (PKs) govern the stress responses.
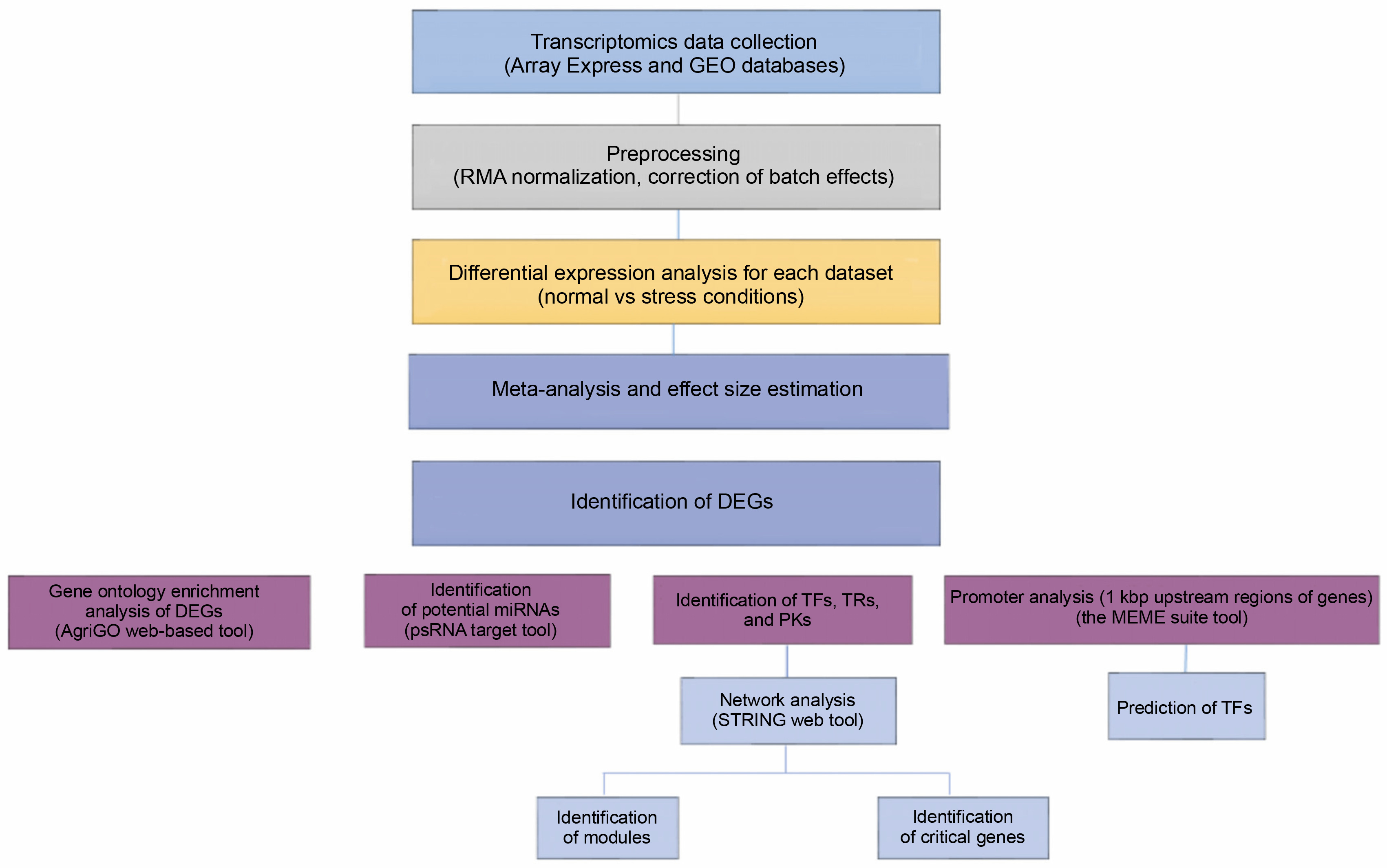
The present study was designed to analyze gene promoters to reveal conserved and enriched motifs with their possible roles in stress response. We used DEGs obtained from the meta-analysis of transcriptomic responses under multiple abiotic stresses in barley. Furthermore, miRNA identification was performed on the DEGs under abiotic stresses. Additionally, regulatory gene sets, TFs, TRs, and PKs families that orchestrate abiotic stress responses were identified through several bioinformatics tools such as PlantTFDB (Tian et al., 2019; Jin et al., 2017) and iTAK (Zheng et al., 2016). Finally, signaling networks were constructed using the TFs, TRs, and PKs, and the key regulators were detected.
Methods
Identification of DEGs
Microarray datasets for abiotic stresses such as drought, cold, salinity, and low-temperature stresses were extracted from Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) – Table 1. All microarray gene expression data were implemented on a single platform (Barley Genome Array, GPL1340).
Table 1
Characteristics of data sets selected for meta-analysis
The probe-gene maps and probe annotation files were downloaded from the Affymetrix site (www.affymetrix.com). The robust multiple average (RMA) method was used to normalize the raw expression data of each array (Irizarry et al. 2003). For this purpose, the Affymetrix Expression Console software was used. When normalization was performed to adjust batch effects by applying the SVA R package (Leek et al., 2012), the empirical Bayes method was used (Johnson et al., 2007).
To find the probe sets that were expressed differentially based on the false discovery rate, a meta-analysis was performed using a nonparametric rank production method (Breitling et al., 2004). A probe set was considered to be differentially expressed if FDR was < 0.01. More analyses were performed on the 3721 founded probe sets. The analyses included determination of TFs and PKs, cis-element analysis, and construction of signaling networks. Overall, 1694 and 2027 probe sets showed significant upregulation and downregulation of expression, respectively. BLASTX from IPK Barley BLAST Server was used to identify the Gene ID accessions related to the probe sets.
Promoter motif analysis of genes corresponding to DEGs
The 1 kbp upstream anking regions of genes corresponding to DEGs were extracted from Ensembl Plants (http://plants.ensembl.org). MEME (Multiple EM for Motif Elicitation) is one of the most widely used tools for identifying novel signals in sets of biological sequences. It includes the discovery of new transcription factor binding sites (Bailey et al., 2006). MEME (meme. nbcr.net/meme/intro.html) was used to discover conserved motifs on the sequences with its default parameters, except for the maximum number of motifs, which was set to 20, with the threshold E-value of < 1e−4 (Bailey et al., 2009). The Tomtom v 5.0.1 tool (http://meme-suite.org/tools/tomtom) was used to eliminate redundant motifs and to de ne known CRE based on the JASPAR CORE 2018 database with the threshold E-value of 0.05 (Gupta et al., 2007; Khan et al., 2017). The GOMo tool (http://meme-suite.org/tools/gomo) was also applied to identify possible biological roles for motifs (Buske et al., 2010).
MAST (Motif Alignment and Search Tool) was used for searching biological sequence databases for sequences containing an occurrence of each motif in a given set of motifs. AME (Analysis of Motif Enrichment) of the MEME suite was used to identify known motifs that are relatively enriched in a set of sequences. To scan given sequences for individual matches to each of the motifs, FIMO (Find Individual Motif Occurrences) of the MEME suite was implemented (McLeay and Bailey, 2010).
Identification of TFs, TRs, and PKs
To identify TFs families and TRs, BLASTX search against the barley transcription factors (http://planttfdb.cbi.pku.edu) with a cutoff value of E ≤ 10−5 was performed on DEG sequences. Moreover, PKs in DEGs were identified from the BLASTX search against the iTAK database (http://bioinfo.bti.cornell.edu/cgi-bin/itak/index.cgi) with a cutoff value of E ≤ 10−5.
Construction of the signaling networks consisting of TFs, TRs, and PKs
The Pathway Studio software (Ariadne Genomics, 2010) was used to construct a network that is established based on the ResNet database in which direct connection is considered between TFs, TRs, and PKs and their neighbors. The downstream term was selected as the directionality of the relationship to detect upstream regulators. The software calculates the local connectivity (node degrees in the network built) and compares them with the connectivity (node degrees in the ResNet database).
Identification of candidate abiotic stress-associated miRNAs
To identify potential miRNAs that might be related to the downregulated DEGs, their sequences were used as queries for the psRNATarget server (http://plantgrn.noble.org/psRNATarget/) with default parameters, except for the maximum expectation, which was set to 3, and compared with the 71 published miRNAs related to Hordium vulgare. Figure 1 shows the schematic procedure of the main steps of this study.
Results
Promoter motif analysis of DEGs
The 1 kbp upstream flanking regions of genes corresponding to DEGs were analyzed to discover conserved motifs and consensus cis-regulatory elements (CREs).
In this regard, 1424 promoter regions corresponding to the upregulated probe sets and 2018 promoter regions corresponding to the downregulated probe sets were retrieved from Ensemble Plants. Twenty significant motifs were detected with lengths ranging from 15 to 50 bp in the promoters of DEGs by using MEME (Suppl. mater. 1). The GOMo analysis for the motifs demonstrated several biological functions. For the upregulated genes, GO motifs were related to kinase activity, monooxygenase activity, plasma membrane, chloroplast envelope, oxygen binding, and protein glycosylation. On the other hand, the downregulated motifs showed monooxygenase activity, tyrosine kinase signaling, nitrogen compound metabolic process, and protein heterodimerization activity.
As observed in motif analysis, the majority of them show a relationship with the APETALA2/ETHYLENE-RESPONSIVE FACTOR (AP2/ERF) domain and C2H2 zinc finger TF family (28% for each). Analysis of motif enrichment revealed that the highest number of TF members belong to the AP2/ERF class: more than 20% of the whole TFs (Table 2). We found 28 and 54 motifs that exist only in the upregulated and downregulated genes, respectively. Table 3 illustrates their functions in abiotic stresses in barley searched through the PLANT-CARE database.
Table 2
The classes of TFs that bind to the related motif
Table 3
Functions related to the detected motifs that were obtained by checking in the PLANTCARE database
Identification of the TFs, TRs, and PKs
DEG sequences were aligned against the PlntTFDB database to identify TFs. We found 35 TFs related to the upregulated genes and 44 TFs related to the downregulated genes, which, respectively, belong to 19 and 23 families of TFs. Four upregulated genes were categorized in the NAC and bZIP class, while in the bHLH family, seven members were downregulated, which was the highest number of downregulated TFs (Table 4). The C2H2 and NF-YA families had just three upregulated members. The MYB-related family had five out of seven and the WRKY family had five out of six downregulated members, which indicates that these TFs families are mostly downregulated in barley under abiotic stresses.
Table 4
TF families identified using BLASTX search against the barley TFs database
TRs family was one of the identified regulatory genes in our study, with 11 upregulated and 24 downregulated members. TRs related to the auxin/indole-3-acetic acid (AUX/IAA) family contains six members, out of which four members were downregulated. The GNAT (GCN5 related N terminal acetyltransferase) family had the highest number of downregulated TRs (Table 5).
Table 5
TR families identified using BLASTX search against the barley TFs database
| TR family | Upregulated | Downregulated |
|---|---|---|
| AUX/IAA | 2 | 4 |
| SWI/SNF-BAF60b | 1 | 1 |
| HMG | 1 | 1 |
| MBF1 | 1 | 0 |
| Others | 5 | 8 |
| Pseudo ARR-B | 1 | 0 |
| GNAT | 0 | 4 |
| SET | 0 | 1 |
| LUG | 0 | 1 |
| TAZ | 0 | 2 |
| mTERF | 0 | 2 |
| Total | 11 | 24 |
Among the DEGs, 18 upregulated and 32 downregulated PK genes were categorized into six PK families. The largest upregulated PK family was CAMK with seven members. RLK-Pelle containing 18 members was the largest among the downregulated PKs (Table 6).
Construction of the signaling networks consisting of the TFs, TRs, and PKs
The connection between TFs, TRs, and PKs and their neighbors was analyzed by network construction using the Pathway Studio software. Table 7 presents the top five genes that contain the highest number of relationships. AP2, a member of the AP2/ERF class of TFs, has 30, the highest number of relationships with other TFs and PKs genes (Table 7). Therefore, this gene can be introduced as one of the most critical TFs in barley involved in abiotic stresses. AP2 has 22 relationship nodes with TFs, one with PK, and one with TR gene. The role of AP2 has been reported previously. To help activate abscisic acid (ABA) and ethylene (ET) dependent and independent stress-responsive genes, many AP2/ERFs respond to ABA and ET (Xie et al., 2019; Hoang et al., 2017). The results showed that HY5 (bZIP TF family member), SHY2 (AUX/IAA TR family member), MYC2 (bHLH TF family member), and LHY (Helix-Turn-Helix TF family member) formed 21–24 nodes with other genes. These genes can be presented as the most important genes of the network. These genes have 6, 8, 6, and 5 nodes with TFs, respectively; 1, 4, 0, and 3 nodes with TRs, respectively; and no relationship with any of the PKs. The remaining genes in the network analysis had less than 17 nodes. The excel format and network image of the genes comprising all relationships and entities are provided as Supplementary materials 2.
Identification of candidate abiotic stress-associated miRNAs
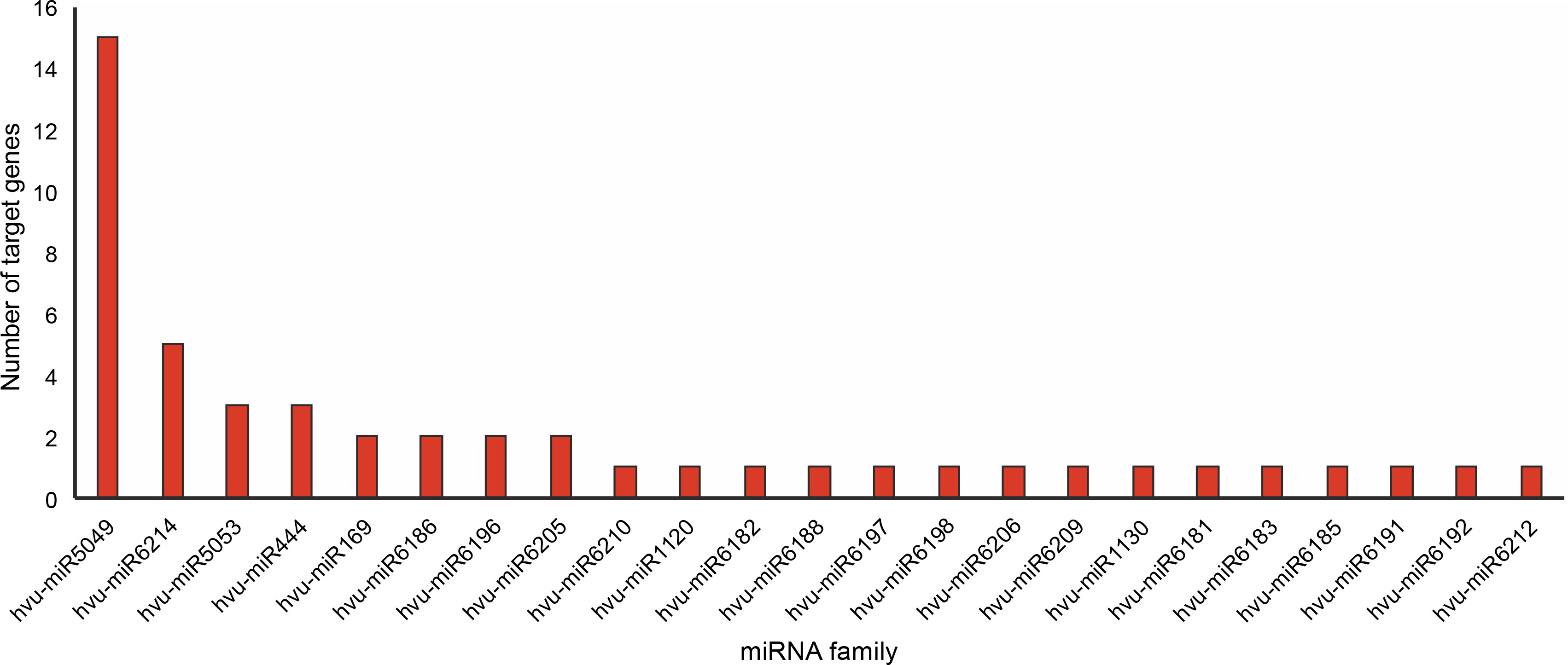
One of the objectives of this study was to identify abiotic stress-associated miRNAs in barley. Hence, the potential miRNAs targeting the downregulated DEGs were predicted using the psRNATarget server. A total of 49 miRNAs belonging to 23 distinct conserved families were found (Fig. 2, Suppl. mater. 3). According to the output psRNATarget server, only five members of the candidate miRNAs participate in translation inhibition, and the remaining are involved in mRNA cleavage processes. The hvu-miR5049 was the largest family with 15 members, followed by the hvu-miR6214 family containing 5 members.
Discussion
Transcriptional regulation plays the main role in the activation and suppression of gene expression. It is controlled mainly by gene promoters and their contributing cis-acting elements and motifs (Zou et al., 2011). According to the results obtained from FIMO, Motif3 was found in the promoters of the highest number (568) of upregulated genes (Suppl. mater. 1). As reported in the motif analysis part, the ERF family transcription factor binds to the Motif3 sequence. This shows that the ERF family transcription factor is the upstream component in a transcriptional cascade that functions as a mediator of the upregulation of gene expression caused by abiotic stress. Previously, it has been indicated that ERF TFs are involved in Arabidopsis (Dubois et al., 2013), rice (Schmidt et al., 2013), and wheat (Zhu et al., 2014) responses to drought, salt, and freezing stresses, respectively. Transgenic Arabidopsis expressing barley ERF TF showed also improved salt tolerance (Jung et al., 2007). Our analysis revealed the participation of the ERF TF family in abiotic stress response in barley.
TFs, TRs, and PKs among the DEGs of barley were identified in this study. It was previously shown that transcription factors such as dehydration-responsive element-binding factors (HvDREB1) and WRKYs (HvWRKY38) are involved in drought stress responses through regulation of the expression of stress-related genes (Gürel et al., 2016). NAC proteins are one of the largest families of TFs that play a role in plant growth and abiotic stress responses. Many studies have shown that overexpression of NAC TFs caused an improvement in stress tolerance such as salt tolerance in Arabidopsis (Jiang and Deyholos, 2006); drought, salinity, and low temperature tolerance in rice (Fang et al., 2008); and dehydration stress tolerance in soybean (Le et al., 2011). The NAC TF family with the highest members among the upregulated genes was also identified in our study. In addition to NAC, the Leucine Zipper (bZIP) family was also identified with the maximum number of members. It was reported that bZIP is one of the largest TFs families that is induced in response to different plant development stages and stress conditions (Pourabed et al., 2015). Both NAC and bZIP transcription factors are known as genes that are induced by the ABA hormone under abiotic stresses (Yoon et al., 2020). Overall, 114 bZIP proteins were reported in H. vulgare, some of which were upregulated under drought and cold conditions (Pourabed et al., 2015). bHLH TFs are the largest family of downregulated genes identified in our study. According to previous reports, nitrogen deficiency caused a decrease in the number of bHLH transcripts in barley (Comadira et al., 2015).
To date, many studies have indicated the role of WRKY TFs in response to cold and drought in barley (Mare et al., 2004), drought in common bean (Wu et al., 2017), and in other crops (reviewed by Bai et al., 2018). The WRKY TF family has 94 members in barley plants (H. vulgare ) (Liu et al., 2014). We identified six differentially expressed WRKY TF family members among barley DEGs, and most of them were downregulated. Although the MYB TF family is large and involved in abiotic stress responses in Arabidopsis (Lippold et al., 2009), rice (El-Kereamy et al., 2012), apple (Cao et al., 2013), and Triticum aestivum (Mao et al., 2011), there is a lack of information regarding the MYB TF family participation in abiotic stress in barley. Our studies revealed that five out of seven members of this family are downregulated.
AUX/IAA plays a role in the growth and development of plants such as wheat (Qiao et al., 2017), Arabidopsis (Kim et al., 2020), and barley (Shi et al., 2020). Our studies indicated that most of the AUX/IAA TR family members were downregulated during abiotic stresses. Nevertheless, the downregulation of ten members of the AUX/IAA family was also reported at the embryogenesis stage in barley following mannitol application (Muñoz Amatriaín et al., 2006).
According to our analysis, the RLK-Pelle PK family members were among the DEGs in barley. To the best of our knowledge, there is only one report on the role of receptor-like kinases (RLKs) in crops under abiotic stress. Yang et al. (2017a) reported RLKs as downregulated kinase genes with negative regulatory effects during drought stress in jute. CaMK is the largest upregulated PK family identified in our study. Several studies have been conducted on the role of calcium-dependent protein kinase (CDPK) in response to abiotic stresses (Yang et al., 2017b). However, there is no report on the role of other CAMK members in the response of barley to abiotic stresses.
The analysis of network connections of the TFs, TRs, and PKs performed in this study showed that HY5 (bZIP TF family member) and LHY (Helix-Turn-Helix TF family member) have a relationship with PHYB in the network, and PHYB has a relationship with short hypocotyl/suppressor of HY2 (SHY2) (AUX/IAA TR family). SHY2 encodes IAA3 and plays a role as a negative regulator of auxin signaling. It connects phytohormone signaling and stress signaling in root development (Li et al., 2020). It shows that HY5 and LHY are upstream genes of SHY2. PHYB belongs to the phytochrome family and plays a role in seed germination and de-etiolation, and it inhibits shade avoidance responses under a high ratio of red : far-red light (R : FR) in Arabidopsis (Reed et al., 1994; Legris et al., 2019).
MicroRNAs (miRNAs), a class of small noncoding RNAs, regulate the expression of genes involved in many different biological processes such as development, environmental adaptation, and stress tolerance by post-transcriptional regulation of gene expression (Alptekin et al., 2017; Parmar et al., 2020; Fan et al., 2020). Manipulation of abiotic stress-associated miRNAs and their targets may pave the way for improving crop performance under several abiotic stresses (Alptekin et al., 2017; Jiang et al., 2019). In the present study, 49 miRNAs belonging to 23 miRNA families were predicted in barley under abiotic stresses. The highest number of miRNAs belong to the conserved family hvu-miR5049 (Fig. 2). Previous studies have reported the role of miR5049, miR169, miR1120, and miR444 in abiotic stresses. For instance, hvu-miR5049 was significantly upregulated under drought conditions in H. vulgare (Hackenberg et al., 2015). miR5049b has been further reported to be upregulated in leaf and root tissues of a drought-tolerant wheat cultivar under drought stress (Akdogan et al., 2015). hvu-miR5049a was upregulated in leaves but downregulated in roots of barley under drought conditions (Hackenberg et al., 2015). The hvumiR169 family, with two members, was one of the abiotic stress-associated miRNAs identified in this study. The miR169 family is found in many different plant species and is considered to be the largest plant miRNA family. Nevertheless, its ubiquitous expression in various plant genera implies its important regulatory effects (Rao et al., 2020). miR169 was shown to be involved in several stresses. For instance, nuclear transcription factor Y (NF-Y), one of the targets of miR169 in several plants such as maize and Arabidopsis, was upregulated in both heat- and cold-stressed wheat (Ni et al., 2013; Gupta et al., 2014; Kumar et al., 2014; Sorin et al., 2014; Luan et al., 2015). It has been shown that the downregulation of miR169 conduces to the transcript accumulation of NFYA5, a critical positive regulator of drought stress resistance in Arabidopsis (Li et al., 2008). Similarly, the decreased expression of miR169 in Arabidopsis was observed by high-throughput sequencing and qRT-PCR analysis under both drought and salt stresses (Pegler et al., 2019). Furthermore, the regulatory role of miR169 and its targets in drought, salt, and cold stress responses in tomato have been recently reported (Rao et al., 2020).
The hvu-miR444 family with three members is another abiotic stress-associated miRNA identified in this study. hvu-miR444b was upregulated in leaves but downregulated in roots under drought conditions in barley (Hackenberg et al., 2015). The same miRNA was also found to be downregulated in response to salinity stress in barley (Deng et al., 2015). hvu-miR444 targets putative MADS-box transcription factor 27 and cryptochrome 1b (Kantar et al., 2010); thus, it controls gene expression related to plant development, morphology, and flowering time.
The miR1120 is yet another abiotic-responsive miRNA identified in this study among the downregulated DEGs in barley under abiotic stress conditions. In accordance with this result, Sinha et al. (2015) reported miR1120 downregulation under N-deficient conditions in wheat (Sinha et al., 2015).
In the present study, the 19 miRNA families involved in barley stress responses have not been reported previously. The members of these miRNA families can be investigated in future studies for revealing their roles in abiotic stresses.
Conclusions
Exploring the abiotic stress-responsive regulatory gene sets in barley in order to understand the mechanism of gene regulatory system under abiotic stress conditions is highly important. For this purpose, TFs, TRs, and PKs families that respond to abiotic stresses in barley were identified based on the analysis of DEGs by bioinformatics tools. Our results also identified new motifs involved in abiotic stress responses. This study sheds light on cis-elements that play a role in response to environmental stimuli in barley. Finally, critical genes and miRNAs were identified as potential candidate genes involved in responses to abiotic stresses.