

Introduction
Diabetic nephropathy (DN) is a severe microvascular complication of diabetes, affecting approximately 39.7% of patients with diabetes, and the global prevalence of DN is 15.48‰ in males and 16.50‰ in females [1, 2]. It is a leading cause of end-stage renal disease worldwide and contributes significantly to the morbidity and mortality of patients with diabetes [3]. The incidence of DN has increased in the last decade, and the mortality of patients with DN is 30 times higher than that of patients with diabetes mellitus without kidney disease, seriously threatening human health [4]. The pathogenesis of DN is complex and involves multiple factors such as hyperglycemia, oxidative stress, and inflammation. Inflammation plays a pivotal role in the initiation and progression of DN [5].
Interleukin-1 receptor-associated kinase 2 (IRAK2) is a member of the IRAK family that regulates various inflammatory signaling pathways [6]. Previous studies have suggested that the IRAK family may contribute to inflammation in patients with diabetes mellitus. Rajaie et al. found that IRAK inhibitors increased insulin sensitivity in animal models [7]. A member of the IRAK family, IRAK1, has been reported to be upregulated in type 2 diabetes mellitus [8] and is involved in the regulation of high glucose-induced endothelial inflammation [9]. IRAK4 is an upstream kinase of IRAK2, and an IRAK4 inhibitor has demonstrated therapeutic effects in DN [10]. However, the specific role of IRAK2 in DN progression and its relationship with inflammation remain unclear.
IRAK2 regulates various signaling pathways, including the nuclear factor-kappa B (NF-κB) signaling pathway [11]. IRAK2 can lead to TRAF6 ubiquitination, which is a key event in NF-κB activation [12]. The NF-κB signaling pathway is a critical cellular signaling pathway that regulates a wide range of biological processes, such as immune responses, inflammation, cell proliferation, and survival [13]. The NF-κB pathway is activated by various stimuli, including pro-inflammatory cytokines, viraland bacterial infections, and cellular stress. Upon activation, the inhibitor of κB (IκB) proteins, which sequester NF-κB in the cytoplasm, are phosphorylated by IκB kinases (IKKs). This phosphorylation leads to the degradation of IκB proteins, allowing NF-κB to translocate to the nucleus and activate the transcription of target genes involved in immune and inflammatory responses [14]. Accumulating evidence suggests that activation of the NF-κB signaling pathway contributes to the pathogenesis of DN. For example, a study by Ren et al. showed that inhibition of the NF-κB pathway protected against renal inflammation and fibrosis in a mouse model of DN [15]. However, the relationship between IRAK2 and the NF-κB signaling pathway in DN has not been thoroughly investigated.
This study aimed to examine the role of IRAK2 in DN progression and its association with inflammation and the NF-κB signaling pathway in a mouse model of DN. Our findings provide new insights into the molecular mechanisms underlying DN and suggest that IRAK2 is a potential therapeutic target for DN treatment.
Material and methods
Materials
Short hairpin RNA (shRNA) targeting IRAK2 (shIRAK2) and a negative control shRNA (sh-NC) were designed using the Designer of Small Interfering RNA website. The sequence of sh-IRAK2 was SS: CAGTATCCTTGTAAGATAA and AS: TTATCTTACAAGGATACTG. Plasmid pcDNA3.1 and pcDNA3.1-NF-κB were constructed by GenePharm.
Animal model
C57BL/6 mice (male, 6-8 weeks old, weighing 20-25 g, n = 48) were obtained from GemPharmatech (Jiangsu, China). Mice were housed in a specific pathogen-free environment at 22 ±2°C, 50 ±10% humidity, and a 12-h light/dark cycle. Mice had ad libitum access to food and water throughout the study period. All animal procedures were approved by the Institutional Animal Care and Use Committee of Xiamen University (XMULAC20220034-8).
Mice were randomly divided into eight groups (n = 6 per group): Control, DN, DN + sh-NC, DN + sh-IRAK2, pcDNA3.1-NC, pcDNA3.1-NF-κB, sh-NC + pcDNA3.1NF-κB, and sh-IRAK2 + pcDNA3.1-NF-κB. DN results from diabetes mellitus type 1, which is commonly induced by streptozotocin (STZ). In this study, DN was induced in mice by a single intraperitoneal injection of STZ (50 mg/kg body weight; Sigma MO, USA) dissolved in citrate buffer (0.1 M, pH 4.5) each day for five consecutive days. The control mice received equal volumes of citrate buffer. Diabetes was confirmed by blood glucose measurements using a glucometer (Roche, Switzerland) at post-injection day three. Mice with blood glucose levels ≥ 16.7 mmol/l were considered diabetic [16]. Model mice were then transfected with sh-IRAK2 or pcDNA3.1-NF-κB according to the grouping.
The body weights of the mice were recorded weekly, and blood glucose levels were detected every four weeks. Mice were euthanized 16 weeks after the STZ injection. Before the mice were euthanized, 24-h urine samples were collected for subsequent experiments. Euthanasia was performed by administering an overdose of sodium pentobarbital (150 mg/kg, intraperitoneal injection), followed by cervical dislocation to ensure death. Kidney tissues were harvested immediately; some of them were fixed in 10% paraformaldehyde for 24 h, while others were snap-frozen in liquid nitrogen, and stored at –80°C for further analyses.
Transfection
For transfection, mice were anesthetized with 50 mg/kg sodium pentobarbital (Sigma, MO, USA) two weeks before the STZ injection. A total of 2 × 109 TU/ml sh-IRAK2 and/or pcDNA3.1-NF-κB lentivirus suspension were injected into mice via the tail vein.
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted from kidney tissues using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s instructions. The concentration and purity of the isolated RNA were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, MA, USA). Total RNA was reverse-transcribed into cDNA using a cDNA Reverse Transcription kit (TIANGEN, Beijing, China) following the manufacturer’s protocol. RT-qPCR was performed using the SYBR Green PCR Master Mix (Lifeint, Xiamen, China) on a StepOnePlus Real-Time PCR System (Agilent Scientific, CA, USA). The PCR cycling conditions were an initial denaturation at 95°C for 3 min, followed by 40 cycles of 95°C for 12 s and 60°C for 40 s. A final melting curve analysis was performed to confirm the specificity of PCR products. The primer sequences for IRAK2 were: forward, 5´-CAGTTCGCTTCCTACGTGAT-3´; reverse, 5´-AGATGTGCTCTCAGGAACCG-3´. GAPDH was used as the internal control, and the primer sequences were: forward, 5´-GGAAAGCTGTGGCGTGAT-3´; reverse, 5´-AAGGT-GGAAGAGTGGGAGT-3´. The relative expression levels of IRAK2 were calculated using the 2-ΔΔCt method.
Western blotting
Kidney tissues were homogenized and lysed in RIPA lysis buffer (Beyotime, Shanghai, China). Protein concentrations were determined using a BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein (25 μg) were separated using SDS-PAGE and transferred to PVDF membranes (Beyotime). The membranes were blocked with 5% non-fat milk in Tris-buffered saline with Tween 20 (TBST) for 1 h at room temperature and incubated with primary antibodies overnight at 4°C. The primary antibodies used were anti-IRAK2 (1 : 1,000; #DF4782, Affinity, MI, USA), anti-p-NF-κB (1 : 1000; #AF2002, Affinity), anti-NF-κB (1 : 1,000; #AF5006, Affinity), anti-p-IKKβ (1 : 1,000; #AF3010, Affinity), anti-IKKβ (1 : 1,000; #AF6009, Affinity), and anti-GAPDH (1 : 5,000; #AF7021, Affinity). After washing with TBST, membranes were incubated with HRP-conjugated secondary antibodies (1 : 5,000; #S0001, Affinity) for 1 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence kit (Thermo Fisher Scientific) and quantified using the ImageJ software (National Institutes of Health, MD, USA).
Periodic acid-Schiff (PAS) staining
Kidney tissues were fixed in 10% paraformaldehyde for 24 h, embedded in paraffin, and sectioned at 5 μm thickness. After deparaffinization and rehydration, the sections were incubated with a 1% periodic acid solution (Beyotime) for 10 min at room temperature, rinsed with distilled water for 5 min, stained with Schiff’s reagent (Beyotime) for 10 min, and incubated with hematoxylin for 3 min. The sections were dehydrated twice using a graded ethanol series (95% for 10 min twice and 100% for 10 min twice), followed by clearing in xylene for 3 min. The glomerular morphological changes were examined under a light microscope (Olympus, Tokyo, Japan).
Enzyme-linked immunosorbent assay (ELISA)
The levels of interleukin (IL)-6, tumor necrosis factor (TNF)-α, TNF-1R, TNF-2R, 24 h urinary protein, urine albumin-creatinine ratio (UACR), and plasma creatinine were measured using specific ELISA kits (Abcam, Cambridge, UK) following the manufacturer’s instructions. Plasma and urine samples were collected from the mice, and centrifuged at 1000 × g for 15 min at 4°C. The supernatants were stored at –80°C until analysis. The assay involved coating 96-well microplates with capture antibodies, washing, blocking, and adding 50 μl of diluted samples or standards. Biotinylated detection antibodies and streptavidin-HRP were sequentially added, followed by 100 μl of TMB substrate solution. The reaction was stopped by adding 50 μl of stop solution, and optical densities were measured at 450 nm using a microplate reader (Bio-Rad Laboratories, CA, USA).
Statistical analysis
Data are presented as mean ± standard deviation (SD). Comparisons between groups were performed using oneway analysis of variance (ANOVA) followed by Tukey’s post-hoc test. Statistical analyses were conducted using GraphPad Prism software (version 8.0; GraphPad Software, CA, USA). P < 0.05 was considered statistically significant.
Results
IRAK2 promotes DN progression and inflammation in mice
IRAK2, a member of the IRAK family, plays a crucial role in various diseases. Our results revealed that IRAK2 expression was elevated in DN model mice compared with control mice (p < 0.01; Fig. 1A, B). Therefore, IRAK2 was knocked down via transfection with sh-IRAK2 to explore its effects on DN progression in mice. After transfection with sh-IRAK2, the mRNA and protein expression levels of IRAK2 were significantly downregulated in the DN model mice (p < 0.01, Fig. 1A and B), indicating successful transfection.
Fig. 1
Interleukin-1 receptor-associated kinase 2 (IRAK2) promotes diabetic nephropathy (DN) progression and inflammation in mice. A) RT-qPCR detected IRAK2 mRNA expression. B) Western blotting detected IRAK2 protein expression. C) Body weight and D) blood glucose levels in mice. E) PAS staining of kidney sections (scale bar = 20 μm)

Weight and blood glucose levels are important indicators of DN progression. During modeling, the weight of the mice was recorded every week, and the blood glucose was determined every four weeks. In the DN model mice the weight was lower (p < 0.01, Fig. 1C) and the blood glucose was higher (p < 0.01, Fig. 1D) than in the control mice. Glomerular morphological changes were detected using PAS staining. The kidneys of control mice had a clear structure and a small colored area (Fig. 1E). Compared with the control group, the glomeruli, thylakoid area, and coloring area of DN mice were enlarged. Shrunk vessels and tubular deformation of tubules were also observed in DN mice. These phenomena indicated that the accumulation of reducing sugars in the glomerular thylakoid intercellular matrix was significantly increased. DN is characterized by kidney damage and declining function. Urinary protein, UACR, and plasma creatinine levels are crucial biomarkers of kidney damage. ELISA results showed that the levels of 24-h urinary protein, UACR, and plasma creatinine were increased in the DN model mice (p < 0.01, Fig. 1F). These results indicated that the DN model was successfully constructed in mice. After IRAK2 knockdown, the body weight of the mice increased (p < 0.05, Fig. 1C), and blood glucose levels decreased (p < 0.01, Fig. 1D). The colored area of the kidney of DN mice was reduced following sh-IRAK2 transfection (Fig. 1E). Our data showed that IRAK2 decreased the levels of 24 h urinary protein, UACR, and plasma creatinine (p < 0.01, Fig. 1F). The above findings revealed a crucial role of IRAK2 in DN progression.
Fig. 1
Cont. E) PAS staining of kidney sections (scale bar = 20 μm). F) Urinary protein, UACR, and plasma creatinine levels were measured by ELISA. G) Inflammatory cytokine (interleukin-6, tumor necrosis factor (TNF)-α, TNF-1R, and TNF-2R) levels were measured by ELISA. DN model mice were generated by intraperitoneal injection of streptozotocin. Data are presented as mean ±SD. *p < 0.05, **p < 0.01 compared with control group, #p < 0.05, ##p < 0.01 compared to DN + sh-NC group
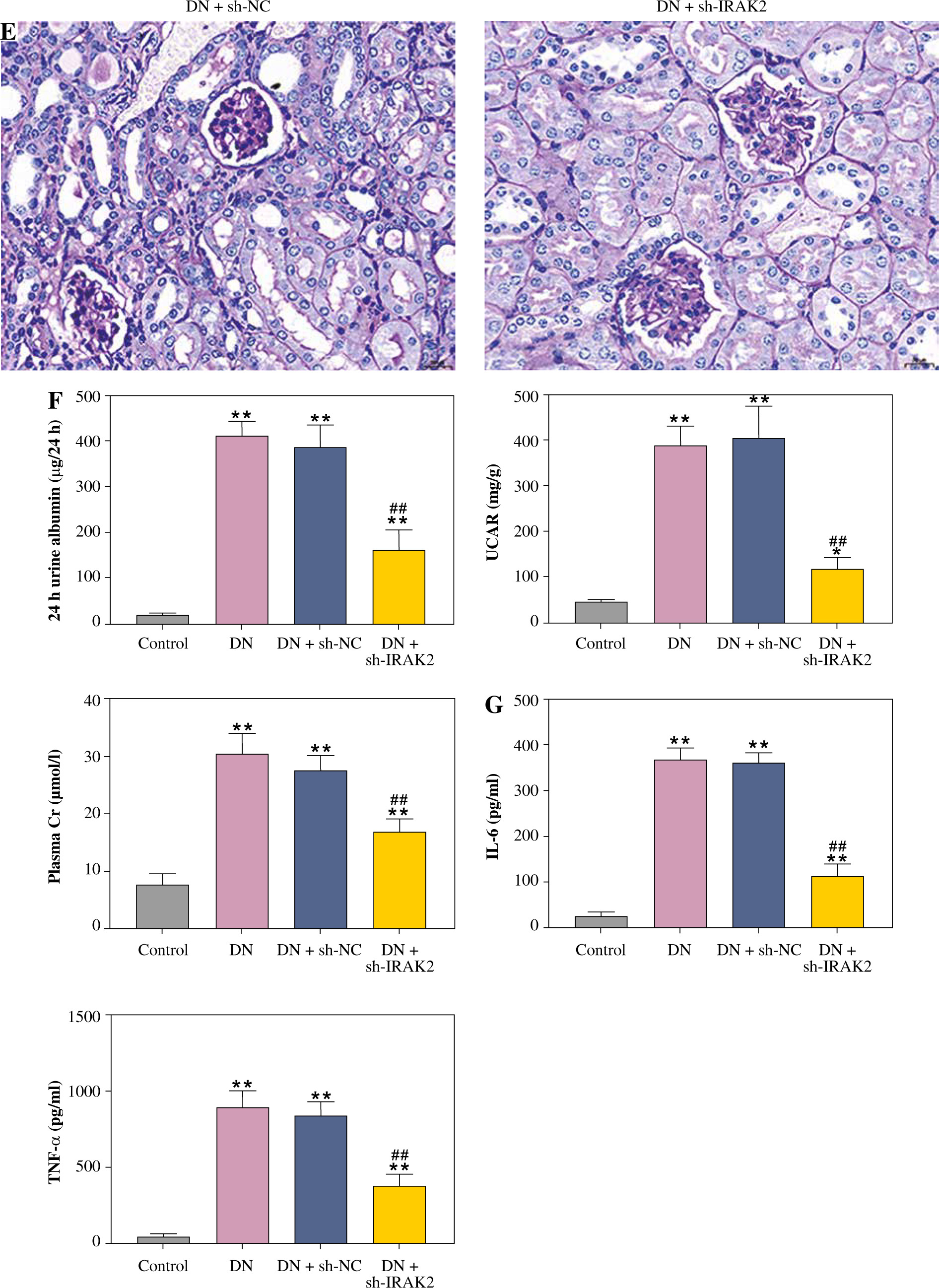
Inflammation is a key factor that contributes to the pathogenesis of DN. We detected the levels of some main inflammatory cytokines (IL-6, TNF-α, TNF-1R, and TNF-2R) associated with DN using ELISA. Compared with the control mice, the levels of IL-6, TNF-α, TNF-1R, and TNF-2R were elevated in the DN mice (p < 0.01, Fig. 1G). IRAK2 downregulation significantly reduced the levels of these inflammatory cytokines (p < 0.01, Fig. 1G).
Fig. 1
Cont. G) Inflammatory cytokine (interleukin-6, tumor necrosis factor (TNF)-α, TNF-1R, and TNF-2R) levels were measured by ELISA. DN model mice were generated by intraperitoneal injection of streptozotocin. Data are presented as mean ±SD.*p < 0.05, **p < 0.01 compared with control group, #p < 0.05, ##p < 0.01 compared to DN + sh-NC group

IRAK2 activates the NF-κB signaling pathway in DN model mice
The NF-κB signaling pathway is a critical cellular signaling pathway that regulates various biological processes, including inflammation. Herein, we investigated the relationship between IRAK2 and NF-κB signaling pathways in DN. In DN model mice, protein expression levels of p-NF-κB/NF-κB and p-IKKβ/IKKβ were upregulated (p < 0.01, Fig. 2). Western blotting results demonstrated that IRAK2 knockdown downregulated the protein levels of p-NF-κB/NF-κB and p-IKKβ/IKKβ (p < 0.01, Fig. 2).
Fig. 2
Interleukin-1 receptor-associated kinase 2 (IRAK2) activates the nuclear factor-kappa B (NF-κB) signaling pathway in diabetic nephropathy (DN) model mice. Protein expression levels of p-NF-κB/NF-κB and p-IKKβ/ IKKβ were determined by western blotting. DN model mice were generated by intraperitoneal injection of streptozotocin. Data are presented as mean ±SD. *p < 0.05, **p < 0.01 compared to control group, #p < 0.05, ##p < 0.01 compared to DN + sh-NC group

Furthermore, the NF-κB signaling pathway was activated by transfecting pcDNA3.1-NF-κB into DN mice. Western blotting results revealed that protein levels of p-NF-κB/NF-κB and p-IKKβ/IKKβ were significantly elevated in pcDNA3.1-NF-κB-transfected mice compared with pcDNA3.1-NC (p < 0.01, Fig. 3A), indicating that pcDNA3.1-NF-κB was successfully transfected into DN mice. In addition, overexpression of NF-κB had no significant effect on IRAK2 expression (Fig. 3B, C). After co-transfection of sh-IRAK2 and pcDNA3.1-NF-κB, the expression of IRAK2 was successfully downregulated in the DN mice (p < 0.05, Fig. 3B and C). Additionally, IRAK2 knockdown reversed the pcDNA3.1-NF-κB-induced activation of the NF-κB signaling pathway, as evidenced by the reduction of p-NF-κB/NF-κB and p-IKKβ/ IKKβ expression after transfection with sh-IRAK2 (p < 0.05, Fig. 3A). These results demonstrated that IRAK2 activates the NF-κB signaling pathway in DN.
Fig. 3
Nuclear factor-kappa B (NF-κB) signaling pathway activation and interleukin-1 receptor-associated kinase 2 (IRAK2) knockdown effects. A) Protein expression levels of p-NF-κB/NF-κB and p-IKKβ/IKKβ was determined by western blotting. B) IRAK2 mRNA and C) protein expression was determined by RT-qPCR and western blotting, respectively. Data are presented as mean ±SD. *p < 0.05, **p < 0.01 compared to pcDNA3.1-NC, #p < 0.05, ##p < 0.01 compared to sh-NC + pcDNA3.1-NF-κB. DN model mice were transfected with sh-IRAK2 and/or pcDNA3.1-NF-κB

IRAK2 accelerates DN progression and inflammation by activating the NF-κB signaling pathway in mice
Subsequently, we explored whether IRAK2 affects DN progression by regulating the NF-κB signaling pathway. DN mice lost weight, and their blood glucose levels were elevated after NF-κB overexpression (p < 0.01, Fig. 4A, B). In comparison with pcDNA3.1-NC, NF-κB overexpression enlarged the glomerulus, thylakoid area, and staining area of the kidneys (Fig. 4C). The levels of urinary protein, UACR, and plasma creatinine were significantly increased after NF-κB overexpression (p < 0.01, Fig. 4D). These results indicated that NF-κB accelerated DN progression; however, IRAK2 knockdown reversed the promoted effects of NF-κB upregulation on DN progression (p < 0.05, Fig. 4A-D). Additionally, NF-κB overexpression elevated the levels of IL-6, TNF-α, TNF-1R, and TNF-2R in the DN mice, which were reduced after IRAK2 knockdown (p < 0.01, Fig. 4D). Based on these findings, we conclude that IRAK2 promotes DN progression by activating the NF-κB signaling pathway.
Fig. 4
Interleukin-1 receptor-associated kinase 2 (IRAK2) accelerates diabetic nephropathy (DN) progression and inflammation by activating the nuclear factor-kappa B (NF-κB) signaling pathway in mice. A) Body weight and B) blood glucose levels in mice. C) PAS staining of kidney sections (scale bar = 20 μm)

Fig. 4
Cont. D) Urinary protein, UACR, and plasma creatinine levels were measured by ELISA. E) Inflammatory cytokine (IL-6, TNF-α, TNF-1R, and TNF-2R) levels were determined by ELISA. Data are presented as mean ±SD. *p < 0.05, **p < 0.01 compared to pcDNA3.1-NC, #p < 0.05, ##p < 0.01 compared to sh-NC + pcDNA3.1- NF-κB. DN model mice were transfected with sh-IRAK2 and/or pcDNA3.1-NF-κB

Discussion
Diabetic nephropathy progression is a multifactorial process, with inflammation playing a central role in its development. IRAK2 is involved in various signaling pathways, including the NF-κB signaling pathway that regulates inflammatory responses. In our study, we found that IRAK2 expression was upregulated in DN model mice, and that IRAK2 knockdown significantly ameliorated kidney damage and inflammatory responses. Importantly, our study demonstrated that IRAK2 downregulation alleviated DN progression by inhibiting the NF-κB signaling pathway.
In recent years, advanced studies on the IRAK family in autoimmune and inflammatory diseases have amplified our understanding of its pivotal roles [17]. IRAK1 expression was elevated in monocytes from patients with type 1 diabetes mellitus [18], and IRAK-M-/- nonobese diabetic mice had a higher incidence of developing diabetes [19]. Immune and inflammatory responses are critical in DN pathogenesis [20]. We observed increased IRAK2 expression in the DN model mice. By knocking down IRAK2, we observed improvements in several DN progression markers, such as body weight, blood glucose levels, and glomerular morphology. Moreover, IRAK2 knockdown reduced the levels of urinary protein, UACR, and plasma creatinine, which are typical biomarkers of renal injury. These results suggest a crucial role for IRAK2 in DN progression and highlight the potential benefits of IRAK2 inhibition for DN treatment.
Increasing evidence has shown that kidney inflammation is crucial for initiating the development and progression of DN. Inflammatory cytokines, including IL-6, TNF-α, and IL-18, are elevated in DN model animals and associated with DN progression [3]. In agreement with previous studies, our results revealed that levels of the inflammatory factors IL-6, TNF-α, TNF-1R, and TNF-2R were increased in the blood of DN model mice. It has been shown that IRAK2 inhibition attenuates lipopolysaccharide-induced inflammatory injury [21, 22]. Our study found that IRAK2 knockdown significantly reduced the levels of pro-inflammatory cytokines, including IL-6, TNF-α, TNF-1R, and TNF-2R. This finding emphasizes the role of IRAK2 in inflammation during DN and supports the hypothesis that targeting IRAK2 may help alleviate inflammation and improve DN outcomes.
IRAK2 is essential in the toll-like receptor and IL-1R signaling pathways, ultimately activating the NF-κB pathway [23]. The IRAK2/NF-κB signaling pathway plays roles in various diseases, such as brain injury [24]. We further explored the relationship between IRAK2 and the NF-κB signaling pathway in DN. Our results showed that IRAK2 knockdown downregulated the protein levels of p-NF-κB/NF-κB and p-IKKβ/IKKβ, suggesting that IRAK2 may activate the NF-κB signaling pathway in DN. This is consistent with previous studies linking the NFκB pathway to DN pathogenesis [25-27]. Furthermore, we demonstrated that NF-κB overexpression worsened DN progression and inflammation, whereas IRAK2 knockdown reversed these effects. These results suggest that IRAK2 promotes DN progression by activating the NF-κB signaling pathway.
Our study has some limitations that warrant further investigation. First, the mouse model may not fully represent human diseases, necessitating additional studies involving human subjects to validate our findings. Second, we focused on the role of IRAK2 in DN progression and its association with inflammation and the NF-κB signaling pathway. Other molecular mechanisms were not examined in this study. Future studies should investigate the relationship between IRAK2 and other pathways that contribute to DN progression.