




In 1966, Davis et al. reported the cases of two young girls presenting with a constellation of symptoms, including chronic dermatitis, joint hypermobility, growth and developmental delay, onychodystrophies, and recurrent infections of the ear, nose, and throat, lungs, and skin, primarily caused by Staphylococcus aureus. They coined the term “Job syndrome” to describe this condition [1]. Six years later, another group of researchers led by Buckley identified similar clinical manifestations in 2 patients. In addition to the previously described symptoms, these patients exhibited facial dysmorphism, fungal infections, increased eosinophils, and elevated levels of immunoglobulin E (IgE) [2]. Subsequently, Clark et al. identified an additional anomaly related to “chemotaxis of neutrophils” [3]. These abnormalities of hyper-IgE and deficiency in neutrophil chemotaxis were also observed in the patients initially described by Davis and his team [4].
Buckley syndrome is a complex genetic disorder, and its most likely mode of transmission is autosomal dominant inheritance with variable penetrance [5–8]. However, it is important to note that many cases of this syndrome appear to manifest sporadically without a clear familial history [9].
The usual diagnostic criteria for Buckley syndrome include: significant elevation of IgE in the blood, typically exceeding 2000 IU/ml [10, 11], moderate increase in the number of eosinophil white blood cells [10, 11], presence of eczematiform dermatitis, and recurrent bacterial infections, particularly those caused by Staphylococcus aureus, affecting the ear, nose and throat, skin, and pulmonary regions and manifesting early in childhood.
In addition to these diagnostic criteria, less common clinical and biological manifestations may be observed in patients with this syndrome. These include vascular anomalies, skeletal abnormalities such as scoliosis, osteopenia, oligoarticular arthritis (affecting 1 to 4 joints without signs of general inflammation), cranial synostosis, as well as facial anomalies, including a prominent forehead and flattening of the base of the nose, and oral anomalies such as delayed tooth eruption and mucosal lesions.
An 11-year-old boy, with a history of atopy, presented 2 weeks before his first hospitalization in the pediatric ward with a cough accompanied by purulent sputum, chest pain evolving in a febrile context, general malaise, and weight loss measured at 8 kg over the past 6 months. Clinical examination revealed dysmorphic facies and coarse features, along with chronic eczematiform lesions. The medical history did not reveal any previous neurological manifestations. The complete blood count showed neutrophil-predominant leukocytosis, and the C-reactive protein (CRP) was elevated at 229 mg/l with a high eosinophil count of 1500/µl.
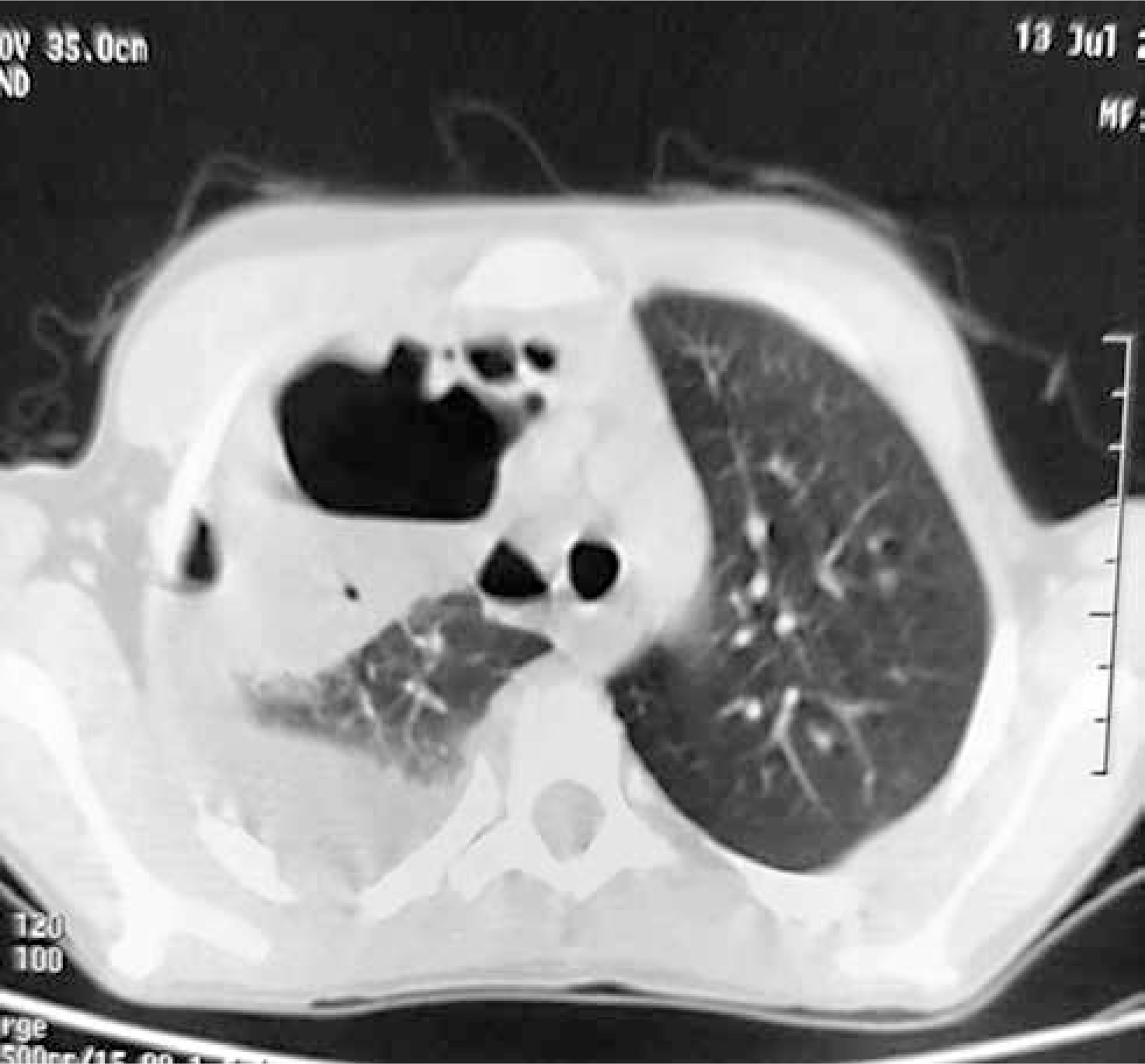
Chest X-ray (Figure 1) revealed features of right hydropyo-pneumothorax, and thoracic CT confirmed the right hydro-pneumothorax associated with abscessed collections in the middle and upper right lobes and subdiaphragmatic region (Figure 2). Bacteriological study of the pus isolated Staphylococcus aureus, while cytobacteriological examination of the sputum was sterile. Acid-fast bacillus (AFB) staining in sputum and X-pert gene testing were negative. HIV serology and hydatid disease serology were also negative. The recommended treatment involved amoxicillin-clavulanic acid for a duration of 2 months, resulting in a favorable clinical outcome.

Figure 2
Chest CT scan showing right hydro-pneumothorax associated with abscessed collections in the middle and upper right lobes
Three months later, the patient returned with an abscessed collection in the right thigh drained at the pediatric ward. An immunological assessment was conducted, revealing an elevated total immunoglobulin E level of 16191 IU/l. The diagnosis of hyper IgE syndrome was established, indicating monthly infusions of polyvalent immunoglobulins.
Hyperimmunoglobulin E syndrome, initially described in 1966 by Davis et al. [12] and further elucidated in 1972 by Buckley [13], is a complex immunological disorder. Its key features include a consistent elevation in immunoglobulin E (IgE) levels and recurrent infections primarily affecting the skin and respiratory tract, often caused by Staphylococcus aureus. This condition manifests early in childhood without gender distinction, and cases diagnosed in adulthood are exceptionally rare. Recurrent infections become apparent as early as 6 months of age, characterized by frequent formation of cutaneous and pulmonary abscesses, as well as the presence of pneumatoceles. Frequent otitis, hepatic abscesses, meningitis, thrombophlebitis, and gingivitis are commonly observed. Patients often present with osteopathies, such as osteoporosis, and bone and joint infections. Atopic dermatitis is frequently evident from the early weeks of life. Atypical facial features, such as hypertelorism and coarse features, are commonly observed [14, 15]. Indeed, our patient exhibited coarse features and a history of dermatitis, and the Buckley syndrome was revealed by pulmonary abscesses and a cutaneous abscess. Staphylococcus aureus is the most frequently implicated infectious agent, although Haemophilus influenzae or Streptococcus pneumoniae may also be associated [15]. IgE levels are significantly elevated, often exceeding 2000 U/ml, while IgG, IgA, and IgM levels remain within normal limits [15, 16]. Allergic manifestations such as eosinophilia, eczema, and increased IgE levels are attributed to an imbalance in the distribution of Th1/Th2 type T lymphocytes, without primary immunodeficiency observed. This disease is of genetic origin, with mutations identified on chromosome 4, following an autosomal dominant mode of transmission, although sporadic cases may occur [17]. Mutations in the gene encoding the STAT3 transcription factor are responsible for hyperimmunoglobulin E, with de novo mutations for sporadic cases and autosomal dominant mutations for familial forms [18]. Mutant proteins inhibit the activity of the protein encoded by the wild-type allele, leading to a significant reduction in STAT3 activity in peripheral blood cells. Two additional underlying mechanisms have been identified, namely a decrease in IgE catabolism and an increase in IgE levels related to recurrent infections [17, 18]. Treatment primarily relies on regular antibiotic prophylaxis, notably trimethoprim-sulfamethoxazole, aimed at improving infections and reducing serum IgE levels. This reinforces the hypothesis that recurrent infections contribute to hyperimmunoglobulin E. Several experimental treatments have been attempted, but the results are mixed (levamisole, ascorbic acid, cimetidine, antihistamines, plasmapheresis). Nevertheless, interferon-γ can normalize neutrophil movement in vitro and inhibit IgE production, while interferon-a can block IgE synthesis by B lymphocytes in vitro. The prognosis of the disease is strongly linked to the severity of pulmonary infections, which can lead to acute respiratory distress, lung tissue destruction, and complications such as bronchiectasis, bronchopleural fistulas, and pneumothorax [15].
In conclusion, Buckley syndrome is a rare immune disorder characterized by recurrent infections, elevated serum IgE levels, and a prognosis linked to the severity of pulmonary involvement.