

Introduction
Sepsis is a disorder of the body’s immune response secondary to severe infections, burns, trauma major surgeries, etc. [1, 2]. The imbalance in the secretion of pro-inflammatory and anti-inflammatory factors is involved in the pathophysiological process of sepsis, which often involves multiple systems and organs, and it is one of the diseases with a high mortality rate [3, 4]. Sepsis often involves the heart, and myocardial injury, including myocardial systolic and diastolic dysfunction and reduced ejection fraction, can be seen in the early stage of sepsis [5]. Septic myocardial dysfunction is one of the reversible myocardial dysfunctions in septic multi-organ dysfunction [6]. Previous investigations have demonstrated that septic myocardial injury is closely associated with oxidative stress, excessive inflammatory reactions, apoptosis, reduced viability, mitochondrial dysfunction, endoplasmic reticulum stress, and focal death [7]. The more central mechanisms include an excessive inflammatory response, oxidative stress, and apoptosis. Therefore, modulation of these functions is important for the treatment of sepsis.
Long-stranded non-coding RNAs (lncRNAs) do not encode proteins per se, and regulate protein-coding gene regions as RNAs with important functional roles in inflammatory responses and rheumatoid arthritis [8]. lncRNA NEAT1, UCA1, and CASC2 partially play an important role in the inflammatory response and oxidative stress induced by lipopolysaccharide (LPS) [9-11]. LncRNA KCNQ1OT1 acts as a factor and is involved in myocardial development and metastasis through regulating viability and apoptosis by sponging miR-192-5p [12]. In the cell and animal models of sepsis, the TTN-AS1/miR-29a axis regulates oxidation, inflammation, and myocardial dysfunction [13]. In osteoarthritis, DLEU1 is involved in the secretion of inflammatory factors interleukin (IL)-6, IL-8, and tumor necrosis factor α (TNF-α) by targeting miR-671-5p, suggesting that DLEU1 contributes to the overexpression of inflammatory factors [14]. In neuropathic pain, the silenced DLEU1 prevented the inflammatory condition of rats with chronic constrictive injury, clarifying that DLEU1 contributes to the inflammatory progression [15]. Sepsis is a manifestation of systemic inflammatory dysregulation; therefore, DLEU1 may be associated with the abnormal manifestations of sepsis.
Herein, mouse cardiomyocyte HL-1 was induced with LPS to mimic a septic cardiomyocyte injury model. The expression level of DLEU1 was regulated and cardiomyocyte viability, apoptosis, inflammation, and oxidation were measured to predict the effect of DLEU1 on myocardial toxicity. A mouse sepsis model was established by cecal ligation and puncture (CLP) to validate the role of DLEU1 on myocardial injury, inflammation, and oxidative stress.
Material and methods
Cell sources and modeling
The HL-1 cardiomyocytes used in the experiments were purchased from ATCC (Manassas, VA, USA), and the cardiomyocytes were cultured in McCoy’s 5A medium (Cienry, Huzhou, China) containing penicillin/streptomycin (1%) and fetal bovine serum (10%), and the cell culture dishes were placed in a cell culture incubator at 37°C, 5% CO2. Gradient concentrations of LPS were added to the medium and the cells were cultured according to a time gradient. Short hairpin RNA targeting DLEU1 (sh-DLEU1) and inhibitors of miR-381-3p were ordered from Thermo Fisher and transfected into HL-1 cells using DOTAP liposomal transfection reagent (Roche Life, Basel, Switzerland).
Ethics statement
Animal experiments were subjected to animal ethical review by Shengli Oilfield Central Hospital. All mouse experiments followed the current international code of ethical use of animals.
Animal modeling and interference
The C57BL/6 mice weighing 25 ±5 g were ordered from The Jackson Laboratory (Bar Harbor, Me, US). The mouse sepsis model was established by cecum ligation and perforation in all mice except the sham group. These mice were anesthetized with isoflurane (3% induced and 1-2% maintained) and then the abdomen was depilated, and a vertical incision of 2-3 cm was made in the midline of the abdomen to open the peritoneum into the abdominal cavity. The distal cecum tissue of the ileocecal valve was ligated with silk thread 1 cm from the end of the cecum, and using an 18-gauge needle, two penetrating punctures were made in the cecum. The peritoneum and skin were closed using 6.0 silk sutures and then resuscitated by intraperitoneal injection of preheated saline (0.05 ml/g). Sham mice underwent dissection only. Five mice were assigned to each group.
Depending on the grouping set, different interfering agents or equivalent measures of saline were injected through the tail vein after modeling.
Determination of cell viability and apoptosis
The Cell Counting Kit-8 (CCK-8) protocol was applied for cell viability detection. 200 ml of cell suspension was added to each experimental well of a 96-well plate. The plate was then gently shaken to ensure even distribution of the cells. The 96-well plate wells were placed in a 37°C incubator for 2 days. In order to avoid light, 100 ml of WST-8 dye [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt] (Boster, Wuhan, China) was added to each well to avoid bubbles during operation. A tin box paper-wrapped 96-well plate was placed in a 37°C incubator to culture for about 1 hour. When the solution in the well plate turned orange, the OD value was read under the 450 nm microplate reader.
HL-1 cardiomyocytes were digested with trypsin without EDTA. After the washing process was repeated twice, the supernatant was discarded, the HL-1 cardiomyocyte suspension was re-suspended, and annexin V-FITC and propidium iodide (Cell Signaling Technology, Danvers, America) were added, 5 ml each, avoiding light. The EP tube was wrapped in tin foil, reacted for 15 minutes, and then placed on a machine to analyze the sample with flow cytometry.
DLEU1 and miR-381-3p in serum and cell culture supernatant
Total RNA was extracted and a Pure high-efficiency blood total RNA extraction kit (TIANGEN, Beijing, China) was used. The concentration of RNA was determined by a continuous wavelength enzyme marker. The RNA A260/A280 ratio of l.8-2.0 suggested that it was qualified. The reverse transcription synthesis of cDNA was carried out after the integrity of RNA was qualified by the test using First Strand cDNA Synthesis (New England Biolabs, Ipswich, US) and miRNA cDNA first chain synthesis kit (TIANGEN, Beijing, China). The sequences of DLEU1 as well as the internal references, GAPDH, miR-381-3p, and U6 primers were designed and synthesized by Sangon (Shanghai, China). The amplification PCR mixing reaction solution was prepared according to the amplification instructions of Luna Universal qPCR Master Mix (New England Biolabs, Ipswich, America), and the fluorescence amplification reaction was performed on the CFX96 system (Bio-Rad, Hercules, CA, USA). The relative mRNA levels were calculated using the 2-ΔΔCT method.
Measurement of cardiac function
Cardiac ultrasound was performed 6 h after modeling to measure left ventricular systolic pressure (LVSP) left ventricular end-diastolic pressure (LVEDP), and maximum rate of left ventricular internal pressure change (± dp/dtmax). After the mice were anesthetized with 2% isoflurane, the hair on the chest was removed and the mice were placed on a thermostat. Changes in left ventricular function in mice were assessed by transthoracic echocardiography using the Vevo770 Echocardiography System (FUJIFILM Visual Sonics; Toronto, Canada).
Identification of inflammatory factors and oxidative stress
All mice were euthanized by intraperitoneal injection of sodium pentobarbital (1%, 100 mg/kg) after post-cardiac ultrasound.
Whole blood was taken from the anterior orbital region of euthanized mice and centrifuged at 1000 × g for 20 min in a serum separator tube and serum specimens were taken. After euthanasia, cardiac tissue was taken from mice to determine the level of oxidative stress. The enzyme-linked immunosorbent assay (ELISA) method was used for subsequent detection. The results were determined using mouse TNF-α, mouse 1β, mouse IL-6, mouse cardiac- type creatine kinase isoenzyme (CK-MB), and a cardiac troponin I (cTnI) kit to test serum specimens. Myocardial tissue homogenate specimens were assayed using mouse superoxide dismutase (SOD), reactive oxygen species (ROS), and mouse malondialdehyde (MDA) kits. All kits were ordered from Jiancheng Bioengineering Research Institute (Nanjing, China).
Validation of targeting relationships
The software LncBook predicted the presence of the binding site of miR-320-3p in the target DLEU1. The wild-type and mutant sequences of DLEU1 were constructed, and the above sequences were inserted into the pmirGLO vector (Promega, WI, United States). The constructed WT-DLEU1 or MUT-DLEU1 and miR-381-3p inhibitors were transfected into DLEU1 wild-type (WT) and mutant (MUT) HL-1 cells. Subsequently, the luciferase fluorescence intensity of each sample was measured using an enzyme marker (Tecan, Switzerland).
Results
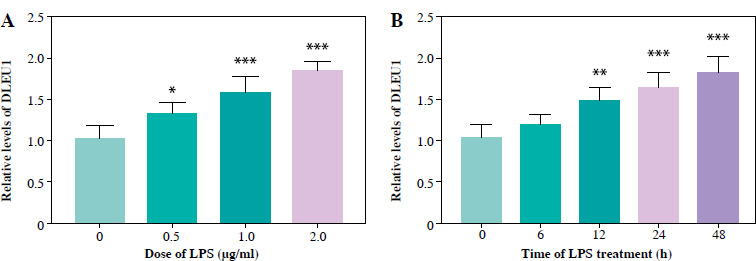
Standardization of LPS dose
After gradient treatment of LPS, we found that the concentration of DLEU1 began to increase at an LPS concentration of 0.5 mg/ml, and at 1 mg/ml the difference in expression was significant (p < 0.05, Fig. 1A). Similarly, when treated for 6 hours, the expression of DLEU1 was not significantly increased, but the difference was significant at 24 hours (Fig. 1B). Therefore, the final scheme of LPS treatment is determined to be 1 mg/ml treatment for 24 hours.
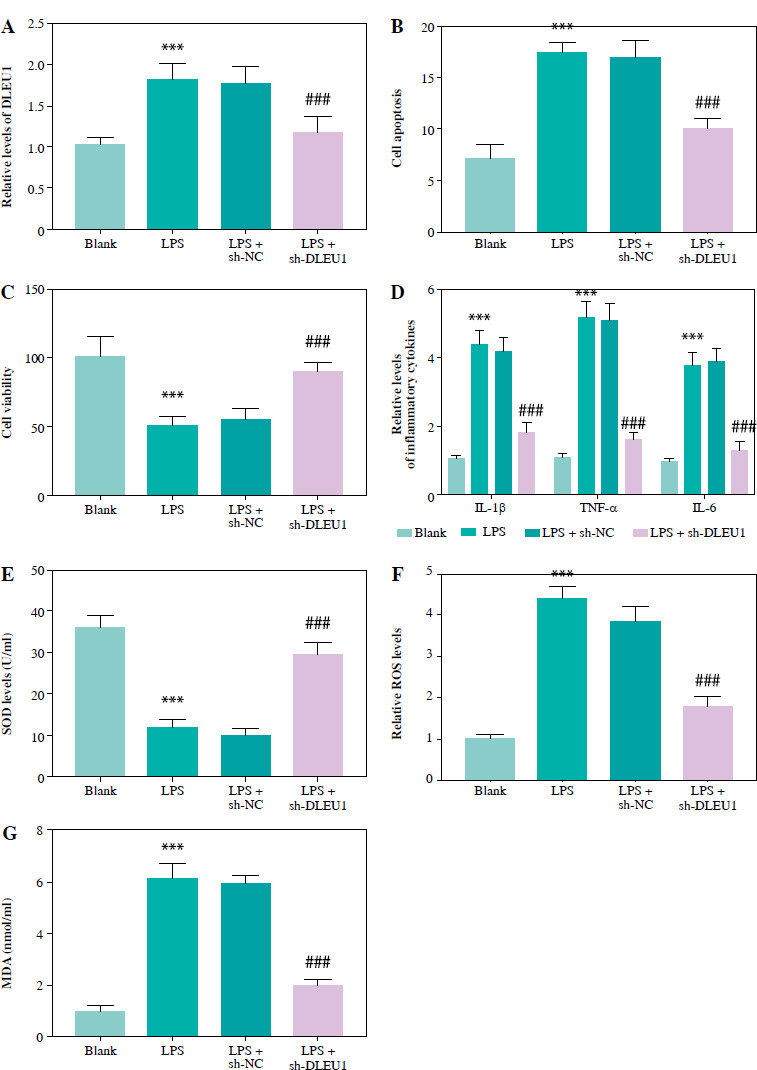
Regulatory effect of DLEU1 on cardiomyocytes
LPS treatment induced up-regulation of DLEU1, and transfection of sh-DLEU1 resulted in partial suppression of DLEU1 expression, indicating that sh-DLEU1 successfully regulated expression (p < 0.001, Fig. 2A). Apoptosis and viability assays showed that DLEU1 interference reversed the effects of LPS on normal cellular functions (p < 0.001, Fig. 2B, C). LPS treatment led to the development of inflammatory responses and oxidative stress, which were attenuated by DLEU1 inhibition (p < 0.001, Fig. 2D-G).
Fig. 2
Role of DLEU1 on HL-1 cells. A) DLEU1 expression levels were upregulated in the LPS group and partially downregulated in LPS + sh-DLEU1. B, C) Downregulation of DLEU1 reversed the LPS-induced elevation of apoptosis and reduction in viability. D-G) Inflammatory factors and oxidative stress indicators are regulated by DLEU1. ***p < 0.001, compared to the blank group; ###p < 0.001, compared to the LPS group
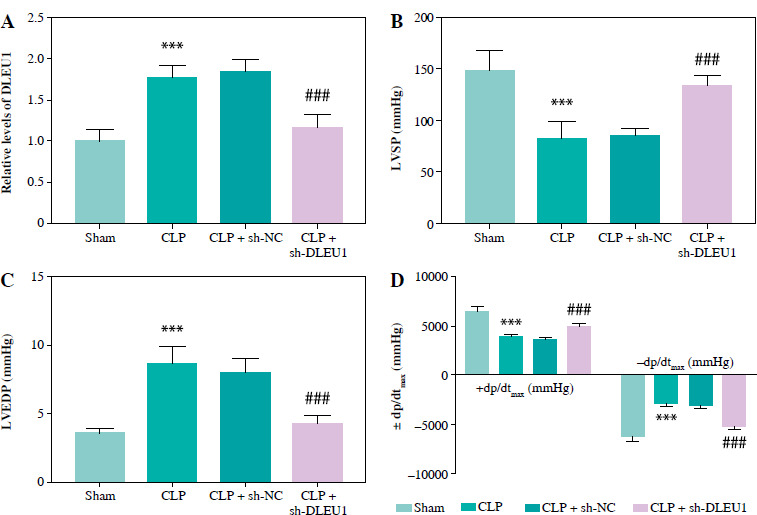
DLEU1 ameliorated myocardial injury of mice
CLP treatment contributed to the elevated expression of DLEU1, which was inhibited by sh-DLEU1 (p < 0.001, Fig. 3A). LVSP, left ventricular end-systolic pressure (LVESP), and ± dp/dtmax indexes reflected the impairment of normal cardiac function by CLP treatment, whereas the reduction of DLEU1 exerted a protective effect (p < 0.001, Fig. 3B-D).
Fig. 3
Role of DLEU1 in myocardial injury. A) sh-DLEU1 successfully inhibited its expression in mice. B-D) LVSP, LVEDP, and ± dp/dtmax were regulated by DLEU1. ***p < 0.001, compared to the sham group; ###p < 0.001, compared to the CLP group
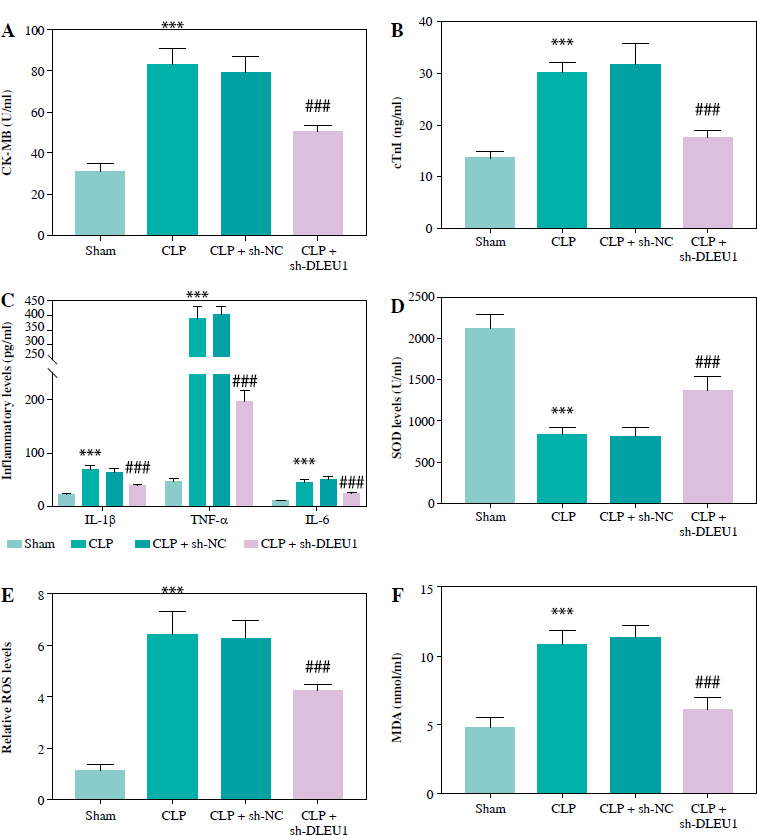
The concentration of CK-MB and cTnI was induced to be abnormally elevated by CLP, and sh-DLEU1 inhibited this aberrant expression (p < 0.001, Fig. 4A, B). The release of inflammatory and oxidative stress factors was promoted by CLP and partially inhibited by sh-DLEU1 (p < 0.001, Fig. 4C-F).
MiR-381-3p was a ceRNA of DLEU1
The predicted targeting sequences of DLEU1 and miR-381-3p are listed in Figure 5A, and the presence of this sequence suggests potential targeting possibilities for both. Reduced expression of miR-381-3p promoted the fluorescence intensity of the WT-DLEU1 group, further confirming our suspicion (p < 0.001, Fig. 5B). The expression of miR-381-3p in the LPS group was dually regulated by LPS and DLEU1, confirming the existence of a targeting relationship between the two (p < 0.001, Fig. 5C).
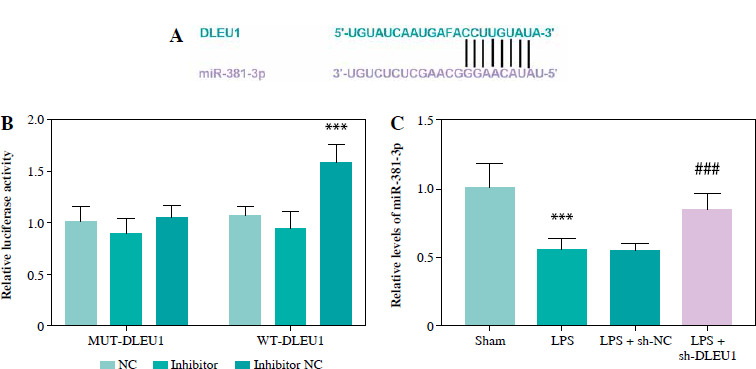
MiR-381-3p impeded DLEU1 effects
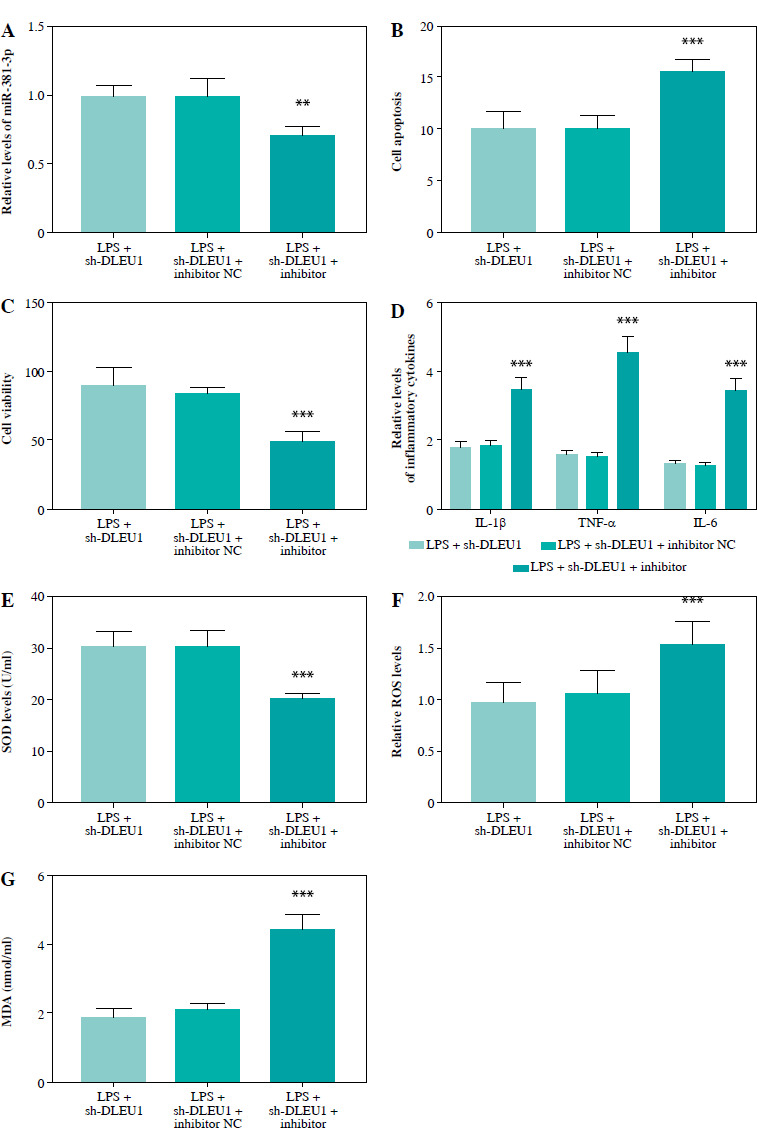
In an in vitro model, miR-381-3p inhibitor could regulate cellular miR-381-3p expression (p < 0.001, Fig. 6A). Reduction of miR-381-3p expression level had a modulatory effect on apoptosis, viability, inflammation, and oxidative stress in the LPS + sh-DLEU1 group (p < 0.001, Fig. 6B-G).
Fig. 6
miR-381-3p was involved in DLEU1 regulation of cardiomyocytes. A) Co-transfection reduced miR-381-3p expression. B-G) Apoptosis, viability, inflammation level, and oxidative stress are regulated by miR-381-3p expression. **p < 0.01, ***p < 0.001
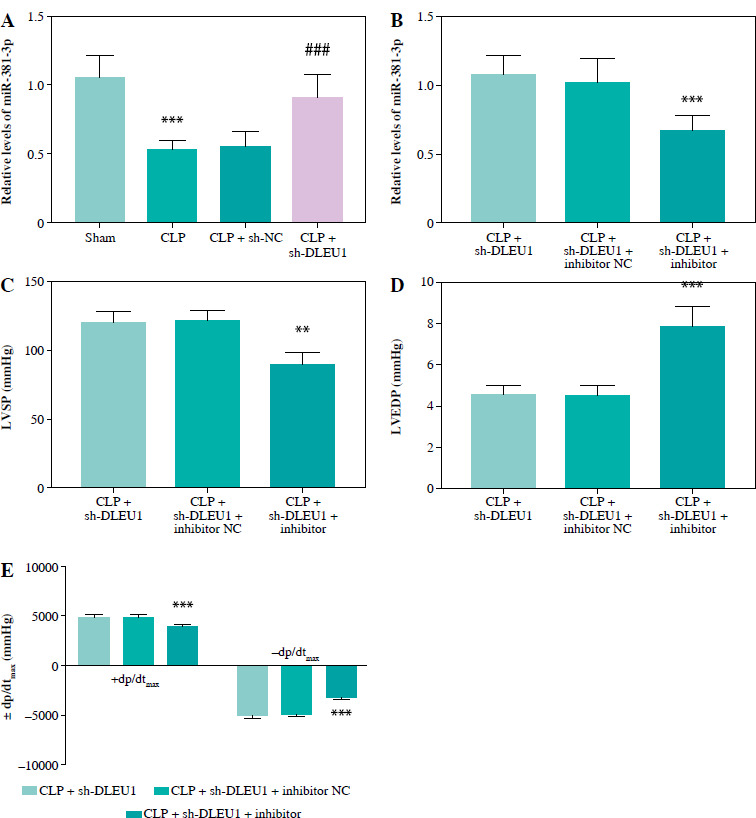
The level of miR-381-3p in the CLP group was reduced, but in the CLP + sh-DLEU1 group the effect was partly reversed (p < 0.001, Fig. 7A). The co-injection of sh-DLEU1 and miR-381-3p inhibitors changed the concentration of miR-381-3p in the CLP + sh-DLEU1 group (p < 0.001, Fig. 7B). MiR-381-3p altered the effects of DLEU1 on the myocardium, and the silenced miR-381-3p reduced LVSP, LVEDP, and ± dp/dtmax (p < 0.01, Fig. 7C-E). As exhibited in Figure 8A-F, CK-MB, cTnI, pro-inflammatory factors, and oxidative stress markers in the CLP + sh-DLEU1 group were regulated by decreased expression levels of miR-381-3p (p < 0.001).
Fig. 7
Regulation of miR-381-3p in a mouse model. A, B) Effects of sh-DLEU1, sh-DLEU1, and miR-381-3p inhibitor on miR-381-3p expression levels in mouse models. C-E) The alteration of miR-381-3p levels changed the LVSP, LVEDP, and ± dp/dtmax levels. **p < 0.01, ***p < 0.001, compared to the sham group or CLP + sh-DLEU1 group; ###p < 0.001, compared to the CLP group
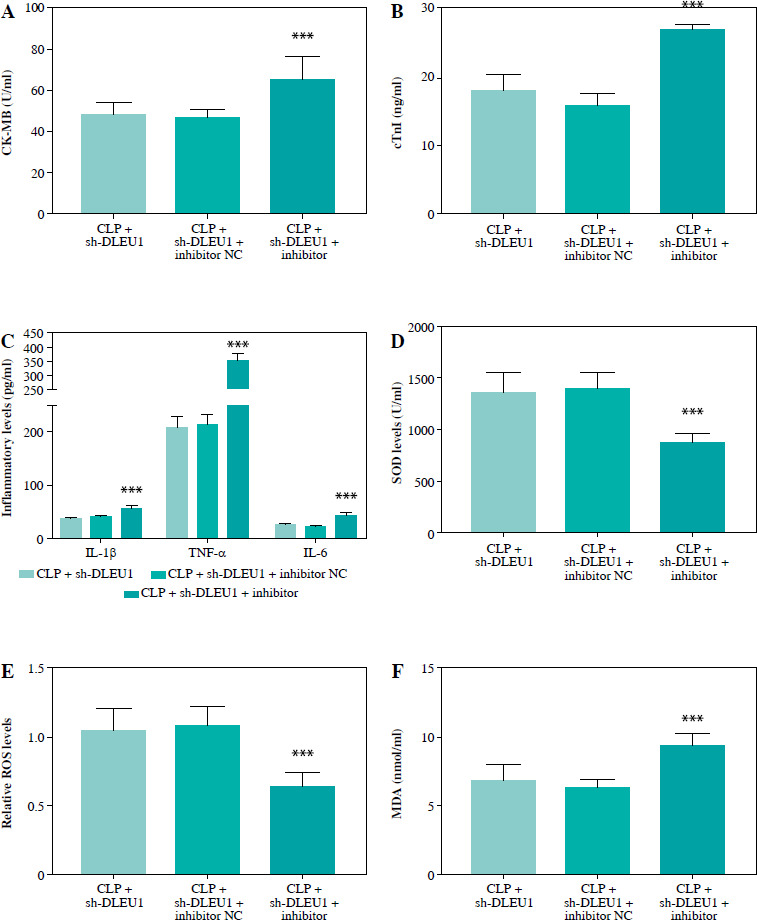
Discussion
Sepsis, caused by infection, can lead to life-threatening organ dysfunction and is one of the leading causes of death in intensive care unit patients [16]. The heart is the main target organ of sepsis, and myocardial dysfunction further promotes the deterioration and development of sepsis, which seriously affects the prognosis of septic patients [17]. However, the exact mechanism by which sepsis causes myocardial damage has not been fully elucidated either. The imbalance between anti-inflammatory and pro-inflammatory responses is the basis for myocardial injury in sepsis [18]. The levels of pro-inflammatory factors such as IL-6, TNF-α, IL-10, and IL-1β are closely related to the severity of sepsis-induced myocardial injury [19]. Oxidative stress in cardiomyocytes promotes protein oxidation and lipid peroxidation, which reduces the contractility of cardiomyocytes, affects cardiac pumping function, and even triggers cardiomyocyte death [20]. Therefore, inflammation and oxidative stress are important for the study of myocardial injury.
LncRNAs participate in the pathophysiological process of sepsis by indirectly regulating the secretion of cytokines [21, 22]. The lncRNA CRNDE and miR-181a-5p regulate the pro-inflammatory cytokines improved by LPS, suggesting that lncRNA can sponge miRNA to change inflammatory situations [23]. The lncRNA NEAT1/miR-370-3p axis mediates the cardiomyocyte damage caused by LPS, indicating that lncRNA may be involved in the mechanism of myocardial injury in sepsis [24]. In this study, the expression of DLU1 increased in a dose-and time-dependent manner, indicating that the concentration of DLU1 rose with both higher LPS concentration and longer treatment time. In cellular experiments, the knockdown of DLEU1 inhibited the effects of LPS on proliferation, apoptosis, oxidative stress, and inflammation, reflecting the protective effects of DLEU1 disruption on cardiomyocytes. The results of animal experiments confirmed the beneficial effects of DLEU1 on oxidative stress, inflammation, and myocardial function in an animal model of myocardial injury. In neuropathic pain caused by osteoarthritis and chronic contraction injury, DLEU1 promotes the release of inflammatory factors, including IL-6, TNF-α, and IL-1β [14, 15]. In the study of diabetic foot ulcer, DLEU1 shows the effect of promoting oxidative stress in cells, which indicates that DLEU1 was related to the occurrence of oxidative stress [25]. All these findings outlined that DLEU1 contributed to the deterioration of myocardial function through regulating cardiomyocytes. However, the cell viability and apoptosis of HL-1 cardiomyocytes were estimated at a single time point. Evaluating cell apoptosis at multiple time points and stages could provide more significant insights and should be considered in future studies. Moreover, other experimental methods are needed for the verification of cell apoptosis.
LncRNAs adsorb miRNA and reduce or even eliminate the influence of miRNA on its target gene. In nasopharyngeal carcinoma and endometrial carcinoma, DLEU1 has been proven to play a biological role by targeting miR-381-3p [26, 27]. This investigation verified that miR-381-3p is a ceRNA of DLEU1, indicating that DLEU1 might exert its effect through sponging miR-381-3p. MiRNA participates in a variety of pathophysiological processes of cardiac diseases. MiR-214-3p, miR-501-5p, and miR-210-3p enhance or alleviate cardiac disorders expedited by sepsis, indicating that these miRNAs are involved in myocardial diseases caused by an uncontrolled inflammatory response [28-30]. This investigation indicated that miR-381-3p mediated the effects of DLEU1 on cells and mouse animal models involved in sepsis by modulating oxidative stress, inflammation, cardiomyocyte damage, and changes in myocardial indices. Liu et al. reported that miR-381-3p inhibits aberrant secretion of inflammation in a cellular model caused by sepsis, suggesting that miR-381-3p is associated with sepsis [31]. Collectively, the DLEU1/miR-381-3p axis is linked to septic myocardial dysfunction.
In summary, the knockdown of DLEU1 provided a positive protective effect on septic cardiomyocytes by inhibiting miR-381-3p and decreasing the level of LPS- induced apoptosis, oxidative stress, and inflammatory response in HL-1 cells. In addition, decreased DLEU1 levels could provide beneficial effects on cardiac function in septic mice by regulating miR-381-3p-mediated myocardial damage and oxidative stress in septic mice.