

Introduction
Inflammatory bowel disease (IBD), a chronic, remitting inflammatory disorder in the gastrointestinal tract, consists of two kinds: Crohn’s disease and ulcerative colitis. Crohn’s disease affects the gastrointestinal tract in a non-continuous manner and causes transmural inflammation, while ulcerative colitis is typified by inflammation in the mucosa and limited to the colon [1, 2]. Although the etiology of IBD remains elusive, it appears to persist in genetically susceptible individuals due to an imbalanced immune response against intestinal microorganisms [3]. Recent evidence suggests that the pathogenesis of IBD results from the interplay of genetic susceptibility and environmental impacts on the microbiome, which, through a weakened intestinal barrier, lead to inappropriate immune activation in the intestines [4]. Inflammatory bowel disease results in a heavy cost to the health-care system [4]. Currently, some drugs targeting interleukin (IL)-23, tumor necrosis factor α (TNF-α), and IL-12 are used in IBD treatment. However, many patients do not respond to these drugs, which have various side effects [5, 6]. Hence, it is important to explore the pathogenesis of IBD and find an effective target for disease treatment.
Long non-coding RNAs (lncRNAs), defined as non-coding transcripts with a length of over 200 nucleotides, are indispensable regulators in the progression of many diseases, such as virus infection, malignant tumors, and IBD. They exert a key regulatory effect on gene expression [7-9]. In accordance with previous genome-wide transcriptome profiling, a great number of lncRNAs are differentially expressed between inflamed and non-inflamed colon pinch biopsies [10]. Some lncRNAs, such as colon cancer-associated transcript-1, have been shown to regulate the development of IBD via binding and inhibiting microRNA (miRNA) expression [9]. Growth arrest specific transcript 5 (GAS5) is an lncRNA that was downregulated in the inflamed biopsies of IBD patients compared to non-inflamed biopsies [11]. Moreover, a significant increase in the level of GAS5 has been identified as the rate-limiting factor in the remission of at least 20% of IBD patients who exhibit resistance to GC treatment [12]. In addition, GAS5 has been reported to regulate intestinal matrix metalloproteinases in pediatric patients with IBD [13]. Whether GAS5 could regulate other key steps in IBD development remains unclear. The micro- RNA miR-23a-3p is overexpressed in plasma samples of patients diagnosed with ulcerative colitis, and its expression is positively correlated with disease severity [14]. The regulatory effect of miR-23a-3p has also been observed in intestinal ischemia/reperfusion rat models [15]. Nevertheless, the functional roles of miR-23a-3p in IBD remain unclear. Blood vessel epicardial substance (BVES) belongs to the Popeye-domain-containing family of tight-junction associated proteins [16]. A recent study demonstrated that the BVES mRNA level was downregulated in human ulcerative colitis biopsy samples, and it may play a protective role in ulcerative colitis and infectious colitis [17]. Furthermore, it is considered a prerequisite for the maintenance of colonic epithelial integrity in experimental colitis by modifying intestinal permeability [17]. However, the involvement of BVES in IBD development has not been explored.
In this study, we analyzed the levels of GAS5, miR-23a -3p, and BVES in colonic mucosal samples of IBD, aiming to reveal the regulatory mechanism of GAS5 in the proliferation, apoptosis, and pro-inflammatory cytokine secretion in an IBD cell model. Our results identified GAS5 as a critical inhibitor of IBD.
Material and methods
Participants
Ten patients with IBD and 10 healthy volunteers were recruited from our hospital in the period from August 2023 to April 2024. The inclusion and exclusion criteria for IBD patients were as follows [18]: 1) aged 18 years or older; 2) the diagnosis of IBD was in accordance with their clinical characteristics, histological results, radiological findings, and endoscopic examination by an experienced doctor. All participants who met the following exclusion criteria were excluded: 1) chronic diarrhea due to other causes; 2) malignant tumors; 3) concurrent autoimmune or endocrine diseases; 4) history of persistent infections; 5) pregnant or breastfeeding women; 6) patients using estrogen for any reason. The IBD group consisted of 6 males and 4 females, with an average age of 34.06 ±3.88 years and a body mass index (BMI) of 22.50 ±1.67. The control group included 5 males and 5 females, with an average age of 45.22 ±8.42 years and a BMI of 24.56 ±4.62. Colonic mucosal tissues were obtained from healthy volunteers and IBD patients during the endoscopy. The study protocol received approval by the Ethics Committee of the First Affiliated Hospital, Anhui University of Science and Technology (approval number: 2023-KY-150-001) and adhere to principles outlined in the Declaration of Helsinki. Written informed consent was collected from all participants.
Cell line
The human fetal colon (FHC) cell line was purchased from ATCC (USA). RPMI-1640 medium (Solarbio, Beijing, China) was used to culture the FHC cells. The FHC cell line was grown in a 5% CO2 incubator at 37°C. For cell culture, RPMI-1640 medium was mixed with 1% streptomycin/penicillin and 10% fetal bovine serum (both from Thermo Fisher Scientific, USA). To induce in vitro inflammation, lipopolysaccharide (LPS; Sigma, USA) at a dose of 1 µg/ml was added to FHC cells and cultured for 24 h [19]. Lipopolysaccharide treatment was applied to all trials, and it was done after transfection. Cells were washed with phosphate-buffered saline three times, each for 10 min, before each use.
Cell transfection
The vectors overexpressing GAS5, vectors overexpressing BVES, empty vector, miR-23a-3p inhibitor, negative control (NC) inhibitor, mimic NC, miR-23a-3p mimic, and vectors expressing short hairpin RNA targeting BVES (sh-BVES) or control short hairpin RNA (sh-NC) (all from GenePharma, China) were delivered into FHC cells utilizing Lipofectamine 3000 (Invitrogen, USA). Transfected cells were incubated 48 h before use in each assay.
MTT assay
The human fetal colon cell viability was quantified using the MTT method. Transfected FHC cells (6 × 103 cells/well) were planted into 96-well plates and subjected to LPS treatment as described above. After 6-72 h, cells were maintained with 10 µl of MTT solution (Solarbio) for 4 hours. Subsequently, 150 µl of DMSO was added and incubated with cells for 10 min to dissolve the violet crystals. Subsequently, the optical density values of cells at 490 nm were detected, and the cell vitality at different time points was analyzed.
Flow cytometric analysis
An Annexin V-FITC/PI apoptosis detection kit (Beyotime, China) was purchased to determine the apoptosis rate of FHC cells. A total of 3 × 105 transfected FHC cells were incubated with Annexin V-FITC and PI (each 5 µl) for 5 min in a light-proof room. Then, the cell apoptotic rate was determined utilizing FlowJo (TreeStar, USA) on a flow cytometer.
Western blotting assay
Protein samples (20 µg each) was separated on 12% SDS-PAGE gels, transferred onto PVDF membranes (Millipore Sigma, USA), and incubated with non-fat milk (5%) for 50 min in an ambient environment. Subsequently, these membranes were maintained at 4°C with anti-BVES (PA5-80386, 1 : 1000, CST, USA) and anti-GAPDH (ab181602, 1 : 1000, Abcam, UK) overnight. Subsequently, the membranes were maintained with a secondary antibody (goat anti-rabbit, ab181662, 1 : 2000, Abcam) at 37°C for 50 min. Finally, an ECL kit (Takara, Japan) was used to visualize the protein bands, and the blots were analyzed using Image J. The expression level of GAPDH was used to normalize that of BVES.
Real-time quantitative PCR (RT-qPCR)
Total RNA was isolated from colonic mucosal tissue specimens and FHC cells by TRIzol reagent (Invitrogen). Subsequently, the cDNA was reverse transcribed from RNA using a cDNA Reverse Transcription Kit (Invitrogen) in accordance with the following conditions: 95°C, 10 min; 42°C, 2 min; 37°C, 15 min; 85°C, 5 s; and then 4°C, 30 min. Subsequently, a SYBR Prime-Script RT-qPCR kit (Takara) was used for RT-qPCR, which was performed on an ABI 7500 system (ABI, USA). The primers in this experiment were as follows: GAS5, 5'-GTGAGGTATGGTGCTGGGTG-3' (F) and 5'-GCCAATGGCTTGAGTTAGGC-3' (R); miR-23a-3p, 5'-GCCGAGATCACATTGCCAGGG-3' (F) and 5'-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAC GGAAAT-3' (R); BVES 5'-CCTCTGCACACAGATCTCCA-3' (F) and 5'-CAAGGCAGCTGATGGACTTT-3' (R). The expression of GAS5 and BVES was normalized to GAPDH (5'-CCAGGTGGTCTCCTCTGA-3' [F] and 5'-GCTGTAGCCAAATCGTTGT-3' [R]). The expression of miR-23a-3p was normalized to U6 (5'-CTCGCTTCGGCAGCACA-3' [F] and 5'- AACGCTTCACGAATTTGCGT-3' [R]). The gene expression levels were quantified by the 2-DDCt method.
Luciferase reporter analysis
The putative binding of miR-23a-3p to GAS5 or the 3' prime untranslated region (3'-UTR) of BVES was verified by luciferase reporter analysis. The mutant (mut) or wide type (wt) sequences of GAS5 or the 3'-UTR of BVES that harbor the binding locations of miR-23a-3p were delivered into the vector pmirGLO to construct the GAS5-wt reporter plasmid, GAS5-mut reporter plasmid, BVES-wt reporter plasmid, and BVES-mut reporter plasmid. Then, these reporter plasmids were transfected with inhibitor NC, miR-23a-3p inhibitor, mimic NC, or miR-23a-3p mimic into FHC cells. At 48 h after cell transfection, the activities of luciferase were examined.
Enzyme-linked immunosorbent assays (ELISA)
The content of proinflammatory cytokines in FHC cell supernatant (1 × 106 per sample) were measured by the human IL-1β, TNF-α, and IL-6 ELISA kits (Beyotime, China). All procedures were in line with the manuals and repeated at least three times.
Statistical analysis
Each experiment was conducted at least three times. GraphPad Prism 5 (USA) was utilized for data analysis. All results were presented as mean ± standard deviation. Student’s t-test was employed for comparing significant differences between two independent groups, while ANOVA (one-way) was applied for multi-group analysis. Pearson’s correlation coefficient determined the relationships between the expression levels of GAS5, miR-23a-3p, and BVES in IBD tissue specimens. P-values lower than 0.05 indicated statistical significance: ***p < 0.001, **p < 0.01, *p < 0.05.
Results
Overexpression of GAS5 promoted FHC cell proliferation, impeded apoptosis and inhibited inflammatory cytokine release
Firstly, GAS5 showed lower expression in the lesion tissues of IBD than in normal tissues (Fig. 1A). Moreover, the GAS5 expression in FHC cells was suppressed by LPS stimulation compared with the control (Fig. 1B). Next, we transfected the GAS5 overexpression plasmid and empty vector into LPS-treated FHC cells. The GAS5 expression was markedly upregulated after GAS5 overexpression plasmid transfection (Fig. 1C). The MTT assay, flow cytometry, and ELISA showed that GAS5 overexpression significantly increased FHC cell viability (Fig. 1D), impeded apoptosis (Fig. 1E), and hindered the release of TNF-α and IL-1β, as well as IL-6 (Fig. 1F). Collectively, these data demonstrate that GAS5 overexpression promotes proliferation and inhibits apoptosis and inflammatory cytokine release in LPS-treated FHC cells.
Fig. 1
Effects of GAS5 overexpression on human fetal colon (FHC) cell proliferation, apoptosis, and inflammatory cytokine release. A) Colonic mucosal samples were collected from ten inflammatory bowel disease (IBD) patients and ten healthy volunteers. Levels of GAS5 were detected by RT-qPCR. B) Expression of GAS5 in FHC cells exposed to LPS was determined utilizing RT-qPCR. C-F) GAS5-overexpressing plasmid and empty vector were transfected into FHC cells. C) Expression of GAS5 in FHC cells was determined by RT-qPCR. D) MTT assay was performed to quantify the viability of FHC cells. E) Cell apoptosis was determined by flow cytometry. F) Release of cytokines was tested utilizing ELISA
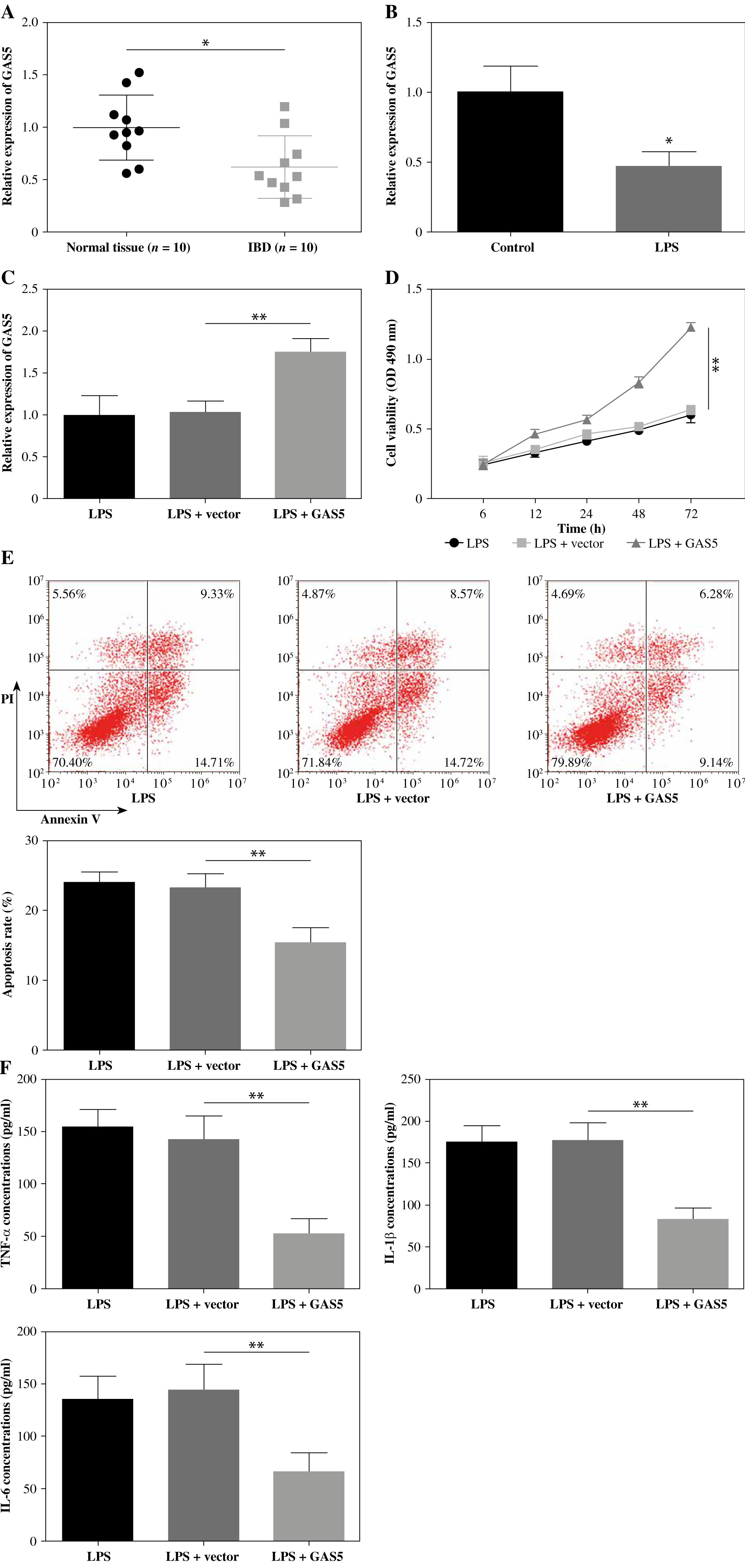
MiR-23a-3p inhibition promoted FHC cell proliferation, impeded apoptosis and inhibited inflammatory cytokine release
MiR-23a-3p was more abundantly expressed in colonic mucosa of IBD patients than in normal colonic mucosal tissues (Fig. 2A). To explore the functional role of miR-23a-3p in FHC cell viability and apoptosis, we transfected miR-23a-3p inhibitor and inhibitor NC into LPS-treated FHC cells. MiR-23a-3p was robustly downregulated by the delivery of inhibitor targeting miR-23a-3p (Fig. 2B). Our findings proved that miR-23a-3p inhibition markedly enhanced FHC cell viability (Fig. 2C), impeded apoptosis (Fig. 2D), and reduced the release of all three inflammatory cytokines (Fig. 2E). These observations suggest that miR-23a-3p inhibition enhances proliferation and inhibits apoptosis and inflammatory cytokine release in LPS-treated FHC cells.
Fig. 2
Impacts of miR-23a-3p inhibition on proliferation, apoptosis, and inflammatory cytokine release in human fetal colon (FHC) cells. A) Colonic mucosal specimens were collected from ten cases of patients with inflammatory bowel disease (IBD) and ten cases of healthy volunteers. Relative expression of miR-23a-3p was measured by RT-qPCR. B-E) FHC cells were treated with miR-23a-3p inhibitor and LPS. B) The level of miR-23a-3p was qualified using RT-qPCR. C) Viability of FHC cells was measured by MTT method. D) Flow cytometry was performed for analysis of cell apoptosis. E) Concentrations of inflammatory cytokines in cell supernatant were analyzed by ELISA
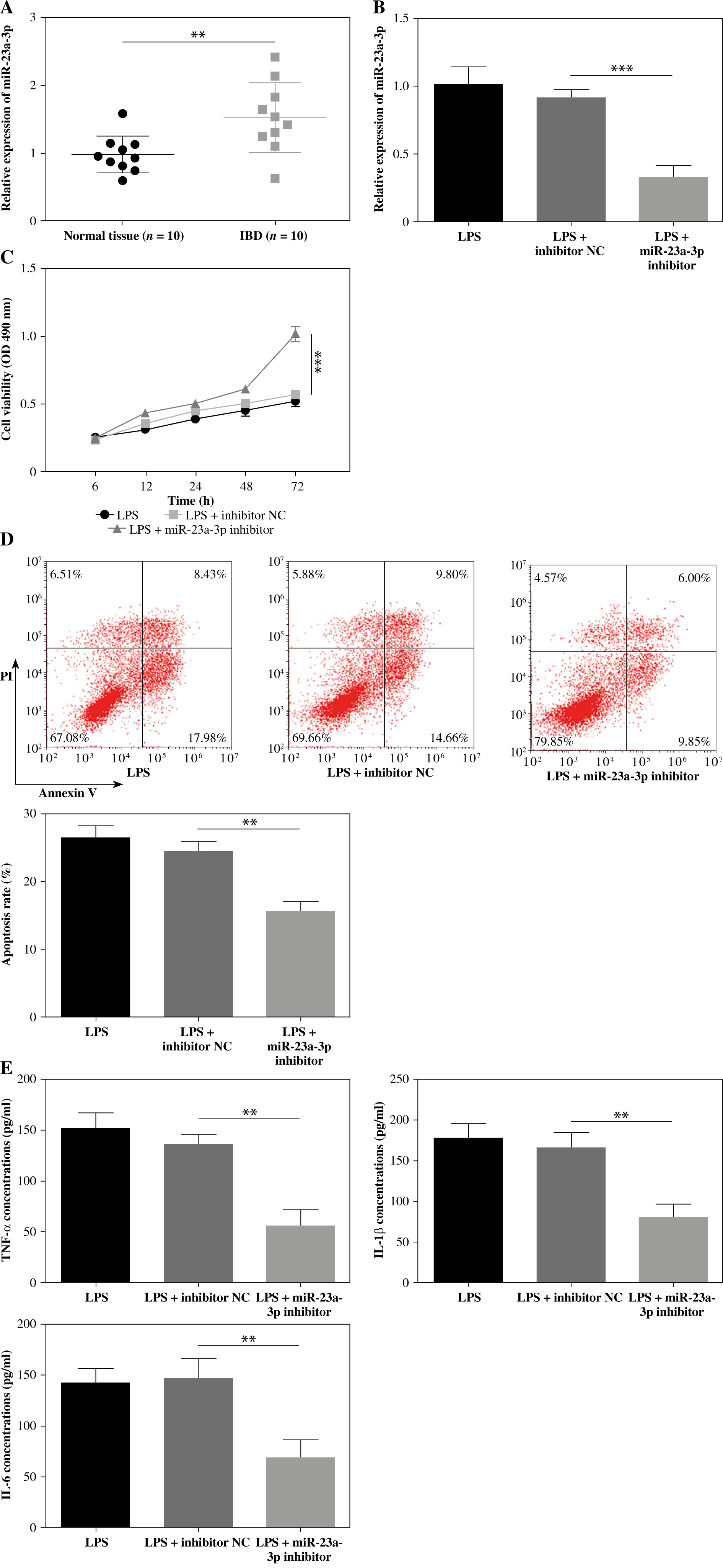
GAS5 targets miR-23a-3p and suppresses its expression
The expression levels of GAS5 and miR-23a-3p in the colonic mucosa of IBD patients were negatively correlated (Fig. 3A). The StarBase database predicted a putative binding location between GAS5 and miR-23a-3p (Fig. 3B). Further analysis revealed that miR-23a-3p mimic effectively reduced the activity of GAS5-wt instead of GAS5-mut. Concomitantly, inhibition of miR-23a-3p notably upregulated the activity of GAS5-wt instead of GAS5-mut (Fig. 3C). In addition, GAS5 overexpression markedly suppressed the miR-23a-3p expression in FHC cells exposed to LPS (Fig. 3D). In summary, GAS5 suppresses miR-23a-3p expression via direct targeting.
Fig. 3
GAS5 targets miR-23a-3p. A) The correlation between GAS5 expression and miR-23a-3p expression in inflammatory bowel disease (IBD) patients was determined by Pearson’s correlation coefficient. B) The binding location between GAS5 and miR-23a-3p was predicted using the StarBase database. C) Binding between miR-23a-3p and GAS5 was confirmed by the luciferase activity assay. D) LPS-treated human fetal colon (FHC) cells were delivered with vectors expressing GAS5 or empty vectors. miR-23a-3p expression was determined by RT-qPCR
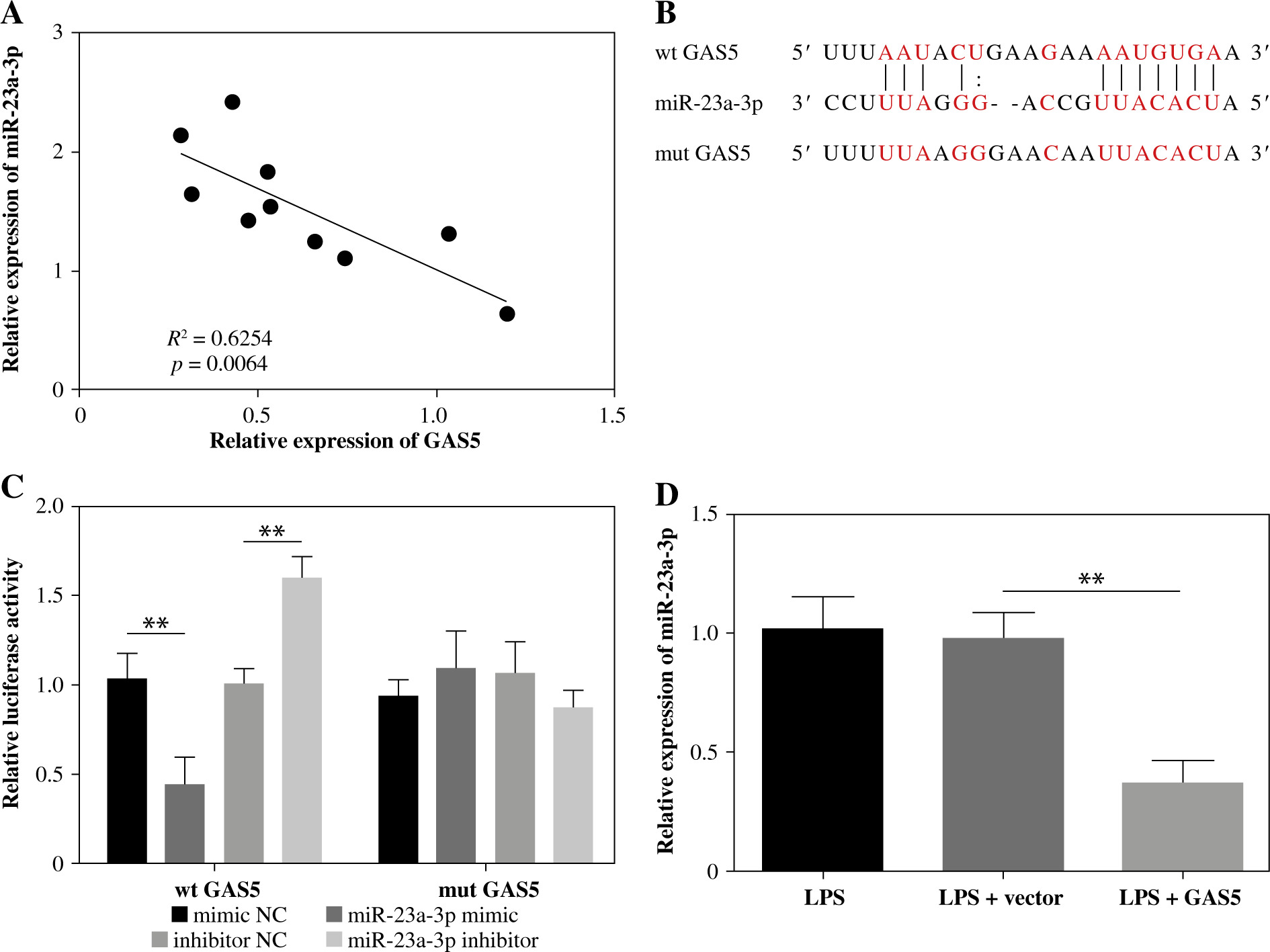
MiR-23a-3p targets BVES and hinders its expression
BVES showed lower expression in IBD colonic mucosa than in healthy tissues (Fig. 4A). The mRNA levels of BVES and miR-23a-3p were negatively correlated in colonic mucosa of IBD patients (Fig. 4B). The TargetScan database predicted a putative binding location between the 3'-UTR of BVES and miR-23a-3p (Fig. 4C). The binding was then proven by the luciferase activity method. The luciferase activity of the BVES-wt group, not the BVES-mut group, was downregulated by increasing miR-23a-3p and upregulated through decreasing miR-23a-3p (Fig. 4D). We further detected the expression of BVES in LPS-exposed FHC cells delivered with inhibitor NC or miR-23a-3p inhibitor. As shown in Figure 4E, F, silencing of miR-23a-3p led to notable upregulation of BVES expression at both gene and protein levels. Overall, BVES is targeted and inhibited by miR-23a-3p.
Fig. 4
BVES is targeted by miR-23a-3p. A) Relative levels of BVES in colonic mucosal samples collected from 10 inflammatory bowel disease (IBD) patients were measured by RT-qPCR. B) The correlation between miR-23a-3p expression and BVES expression in IBD patients was determined by Pearson’s correlation coefficient. C) Binding sequences between the 3'-UTR of BVES and miR-23a-3p were analyzed utilizing the TargetScan database. D) Binding between them was confirmed by the luciferase activity method. E, F) BVES mRNA and BVES protein expression in the LPS-treated FHC cells treated with inhibitor NC or miR-23a-3p inhibitor was analyzed by RT-qPCR and Western blotting, respectively
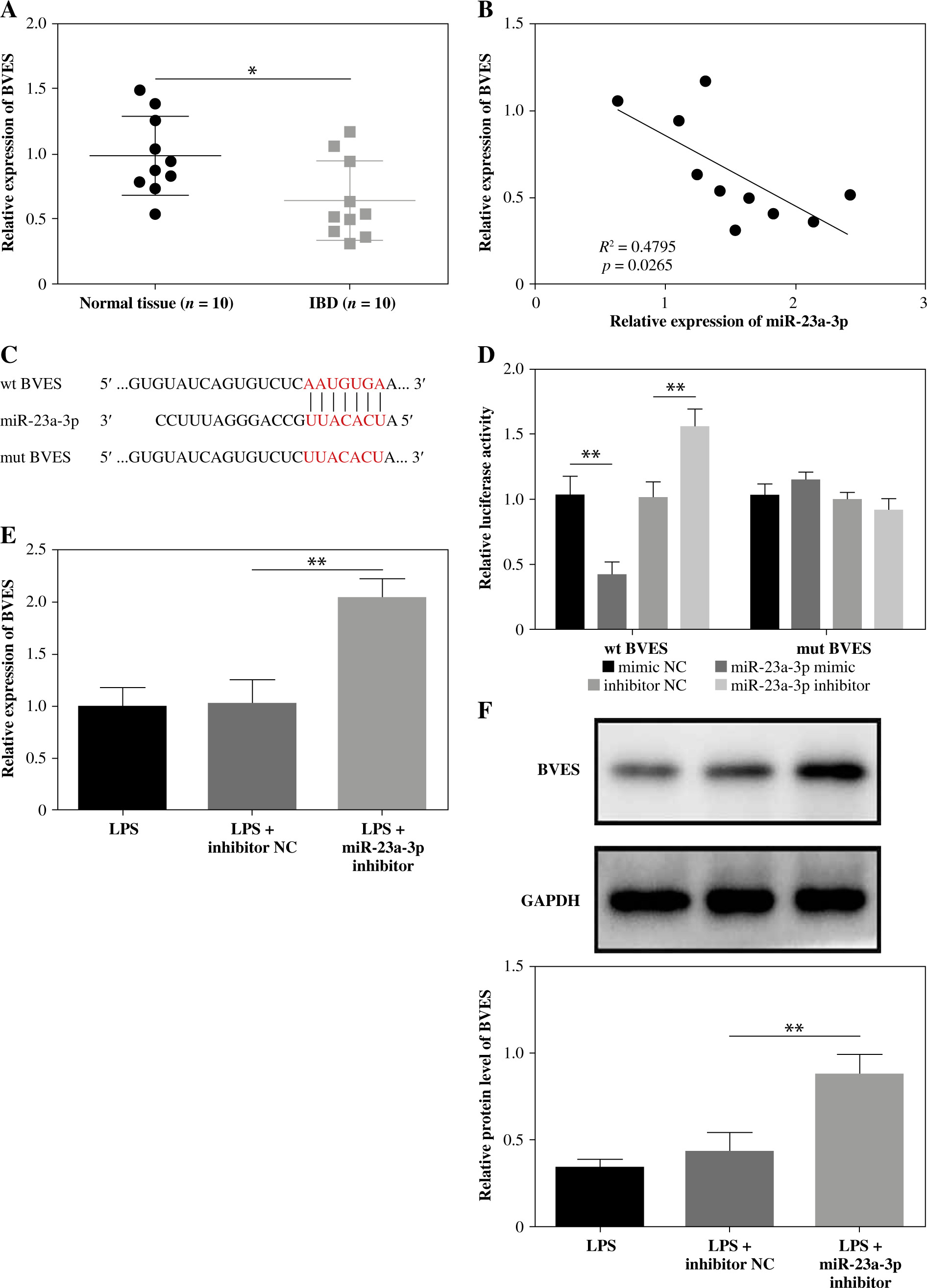
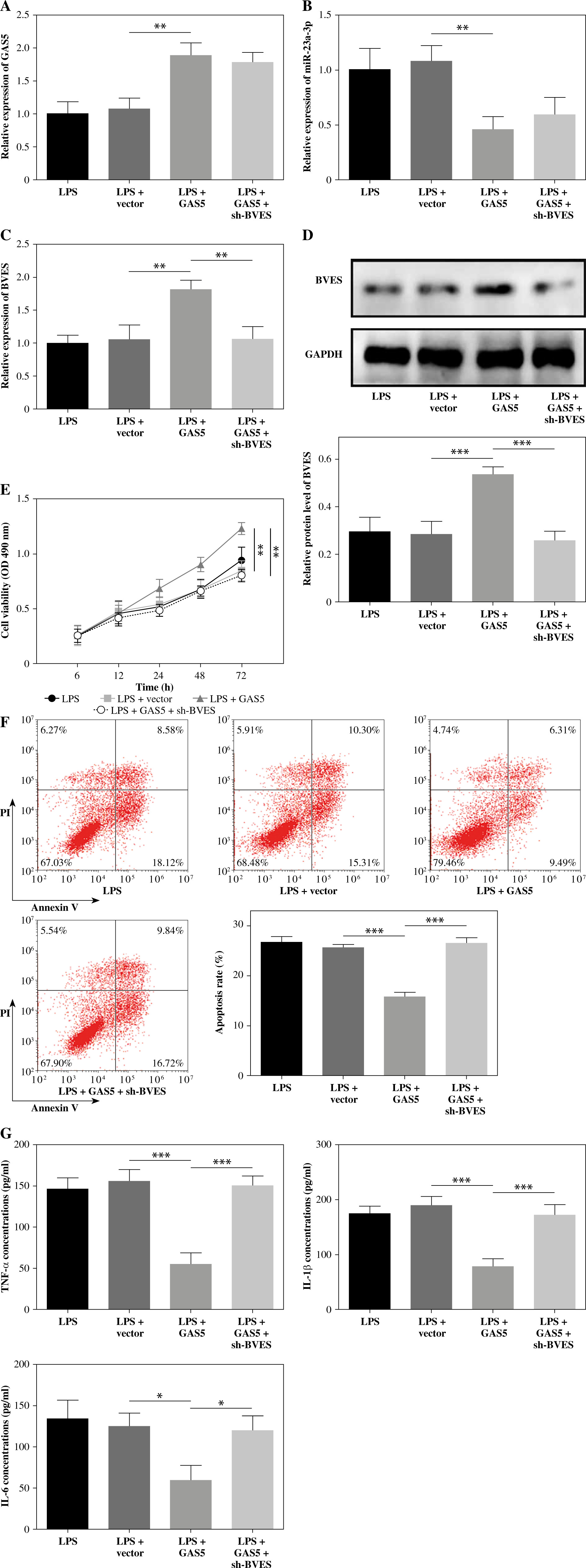
GAS5 promotes proliferation and inhibits apoptosis and inflammatory cytokine release by upregulating BVES
Lastly, we used vectors expressing BVES shRNA to transfect LPS-treated FHC cells delivered with the GAS5-overexpressing plasmid or empty vector. The expression of GAS5 was notably promoted, whereas miR-23a-3p was hindered by the transfection of GAS5-overexpressed plasmid (Fig. 5A, B). Meanwhile, both BVES protein and BVES mRNA were upregulated after GAS5 overexpression plasmid transfection, which were partly reversed after sh-BVES transfection (Fig. 5C, D). Importantly, our data suggested that GAS5’s impacts on FHC cell viability and apoptosis were partly rescued by BVES silencing (Fig. 5E, F). The inhibition of cytokine production by GAS5 overexpression in LPS-treated FHC cells was also reversed by BVES silencing (Fig. 5G). Overall, GAS5 promoted FHC cell viability, and inhibited cell apoptosis and inflammation through upregulating BVES expression.
Fig. 5
GAS5 regulated human fetal colon (FHC) cell viability and apoptosis, and the production of cytokines via targeting BVES. LPS-treated FHC cells were transfected with vectors expressing BVES shRNA and GAS5-overexpressing vectors. Levels of A) GAS5, B) miR-23a-3p, and C) BVES were measured through RT-qPCR. D) The protein level of BVES was examined by Western blotting assay. E) The MTT method was used to assess the viability of FHC cells
Discussion
Increasing evidence has suggested that lncRNA is a potential target for the therapy of many disorders. Here, we observed downregulated expression of GAS5 and BVES and upregulation of miR-23a-3p in IBD colonic mucosal tissue specimens. Moreover, increased GAS5 expression facilitated the proliferation, suppressed the apoptosis, and reduced pro-inflammatory cytokine production of FHC cells exposed to LPS via inhibiting miR-23a-3p and promoting BVES expression.
LncRNAs do not encode peptides or proteins but have two- and three-dimensional structure, which is different from other non-coding RNAs, such as miRNAs, circular RNAs, and small interfering RNAs [20]. LncRNAs are crucial regulators of gene expression involved in multiple biological process, such as cell proliferation, cell cycle, invasion, and drug resistance [21]. LncRNAs act as endogenous inhibitors, also known as spongers, of miRNA in cells to prevent the target miRNA from binding to its downstream targets, thereby promoting expression of this targets [22]. Nie et al. reported that lncRNA ITSN1-2 was increased in IBD intestinal mucosal tissues, and participated in activation and differentiation of T cells through facilitating IL-23R expression via acting as a miR-125a sponge [23]. Yang et al. reported that lncRNA CRNDE promoted dextran sulfate sodium-induced apoptosis of a colonic epithelial cell line via sponging miR-495 and increasing the level of the suppressor of cytokine signaling 1 [24]. GAS5 has been reported to exert an anti-inflammatory effect in sepsis [25]. In addition, administration of GAS5 decreased neuroinflammation in aged mice [26]. Moreover, GAS5 suppressed inflammatory responses and apoptosis of alveolar epithelial cells in mice with LPS-induced acute lung injury [27]. The present study showed that GAS5 was decreased in the colonic mucosal samples of IBD patients. Increased expression of GAS5 improved FHC cell viability, impeded apoptosis, and inhibited inflammation of FHC cells exposed to LPS. These findings underscore the potential of GAS5 as a promising therapeutic target for IBD and provide a reference for developing novel therapeutic interventions targeting GAS5.
GAS5 has been proven to regulate the development of human diseases through acting as a miRNA sponge in some pathological conditions. For instance, GAS5 exacerbated myocardial depression in septic mice via serving as a miR-449b sponge and subsequently repressing high-mobility group box 1 expression [28]. GAS5 also attenuated cell viability and inhibited cell autophagy by targeting miR-23a/ATG3 expression [29]. Furthermore, Gao et al. observed that GAS5 targeted miR-23a-3p and Toll-like receptor 4 in septic conditions [30]. Liu et al. also found that GAS5 suppressed the proliferation and invasion of osteosarcoma cells via targeting miR-23a-3p and the PI3K/AKT pathway [31]. Here, our experiments showed that GAS5 bound with miR-23a-3p and decreased its level in LPS-treated FHC cells. In addition, miR-23a-3p was increased in IBD pathological conditions. Blockade of miR-23a-3p enhanced the viability of LPS-treated FHC cells and impeded their apoptosis and inflammation. Whether other factors are involved in the GAS5-miR-23a-3p network during IBD development warrants further exploration.
Intestinal epithelial cell apoptosis is a major pathological feature of IBD. It is important for IBD improvement to inhibit the abnormal apoptosis in intestinal epithelial cells [32]. BVES, also named as POPDC1, is expressed in cardiac tissue, skeletal tissue, and the gastrointestinal epithelium, contributing to the maintenance of epithelial integrity [33]. BVES is required for maintaining colonic epithelial integrity in colitis [17], and mice lacking BVES exhibited worse intestinal injury and inflammation [16]. Additionally, BVES regulated intestinal stem cell programs and intestinal crypt viability after radiation [34]. BVES has also been shown to be regulated by BVES antisense RNA 1, inhibiting the proliferation, migration, and invasion of colon adenocarcinoma cells [35]. Here, our results indicated that BVES was decreased in the colonic mucosal samples of IBD patients, and it was a downstream gene of miR-23a-3p. Silencing of BVES notably reversed the impact of GAS5 on the proliferative process, inflammation, and apoptosis of FHC cells exposed to LPS. The identification of the regulatory roles of GAS5 and its downstream targets suggests their potential utility as biomarkers for developing diagnostic tools aimed at early detection and personalized management of IBD.
There are some limitations of the present study. First, we only explored the effects of GAS5, miR-23a-3p, and BVES, and their interactions in vitro. These findings need to be validated with a larger sample size and in animal models. Second, the functions of the “Full-length” (FL) and “Clone 2” (C2) variants of GAS5 were not explored in this study. FL has been shown to considerably enhance cell proliferation by rescuing cell cycle arrest, while C2 is responsible for driving apoptosis [36]. Further investigations on the distinct roles of the two variants are warranted.
Conclusions
In conclusion, our findings highlight the pivotal role of GAS5 in enhancing the viability and suppressing apoptosis and inflammation in FHC cells through upregulation of BVES via miR-23a-3p binding. These results may provide a foundation for future research aimed at translating these findings into clinical applications that could improve outcomes for patients with IBD.