










Introduction
Diabetes is reported to be the most common endocrine disorder (Cho et al., 2018). It is a heterogeneous group of metabolic disorders of carbohydrates, fats, and proteins characterized by chronic hyperglycemia and glucosuria with secondary disturbance of protein and fat metabolism (Dastjerdi et al., 2015). The global prevalence of diabetes has increased tremendously despite the wide array of interventions and treatment options currently used in managing the condition (Wu et al., 2014). This has refocused research interest to exploit plants and natural products for treating and managing patients with diabetes. Plants and plant-derived products may provide a comparative advantage by virtue of their diverse bioactive secondary metabolites and multimodal mechanisms of action (Ononamadu et al., 2019). Furthermore, they can serve as sources of lead compounds that can be optimized for obtaining more active compounds by using synthetic approach.
Postprandial glucose spike and insulin resistance are critical events that characterize the progression and complications of diabetes mellitus (Hossain et al., 2016; Leahy et al., 2019). Thus, it is becoming increasingly popular to target carbolytic enzymes (amylase and glucosidase) and proteins implicated in the insulin signaling pathway (such as insulin receptor (in vitro, in vivo, and in silico)) to provide more insights on the hypoglycemic mechanisms of different plant parts and their constituents (Hossain et al., 2016; Riyaphan et al., 2018).
One of the challenges of contemporary ethno-pharmacological research is the complex nature of plant extracts. Plant extracts are usually composed of a vast array of bioactive principles, of which some may constitute a confounder in the overall activity of the extracts. This could be through interference with the efficacy of the active compounds in treating disease conditions or causing undesired side effects or toxicity due to the presence of certain compound(s) in plant extracts (Chikezie et al., 2015). Nonetheless, the advent of more sophisticated analytical techniques such as high-performance liquid chromatography–nuclear magnetic resonance (LC-NMR), gas chromatography-nuclear magnetic resonance (GC-NMR) high-performance liquid chromatography-mass spectroscopy (LC-MS), gas chromatography-mass spectroscopy (GC-MS), thin layer chromatography-mass spectroscopy (TLC-MS), and computer-aided drug screening and optimization methods is beginning to revolutionize ethno-pharmacological studies. Complex constituents or mixtures used in studies can be resolved into components, identified (using hyphenated techniques of chromatography and mass spectrometry or chromatography and Nuclear magnetic resonance spectroscopy ), validated, and optimized for activity or further studies (through in silico methods) almost simultaneously (Ahmed, 2014).
Gymnema sylvestre R. Br. Is a perennial, woody climber belonging to the family Asclepiadaceae or the “milk weed” family. The genus consists of 40 species, some of which including Gymnema sylvestre, Gymnema montanum, Gymnema yunnanense, and Gymnema inodorum are known for their medicinal properties (Tiwari et al., 2014). G. sylvestre is a slow-growing herb found ideally in tropical and subtropical humid climate and is common in hills of evergreen forests (Tiwari et al., 2014). Extracts of G. sylvestre leaves have been shown to reduce hyperglycemia in laboratory animals, and it is used ethno-pharmacologically to treat adult onset diabetes mellitus (NIDDM). This effect is achieved majorly through stimulation of the pancreas and increase in insulin release, although some other mechanisms have also been suggested (Persaud et al., 1999; Kanetkar, 2007). It is also reported to be effective against arthritis, diuresis, anemia, osteoporosis, hypercholesterolemia, cardiopathy, asthma, constipation, microbial infections, indigestion, and inflammation (Kanetkar et al., 2007; Khan et al., 2019).
Combretum micranthum Fam. (Géézà in Hausa) belongs to the family Combretaceae. It is an undomesticated shrub species commonly found in the Tiger bush region of western Africa on cultivated and fallow grounds; it occurs throughout the continent, but appears to be dominant in sub-Saharan Africa, from Sudan to Nigeria and from Gambia to Congo, with higher occurrence in Senegal, Mali, and Burkina Faso (Iwu, 2014). It is a widely known ethno-medicinal plant used in West Africa (including northwestern Nigeria) for treating several medical conditions. C. micranthum is commonly used for treating diuresis and digestion issues, including gastrointestinal problems, colic, and vomiting, in which a beverage is brewed from the dried leaves (Iwu, 1992; Welch et al., 2017). The control of blood glucose in diabetes is also an interesting effect of drinking C. micranthum tea (Welch et al., 2017). The ethanol extract of the leaves is reported to be rich in polyphenols (tannins, flavonoids, and other components) known to possess various beneficial pharmacological properties such as antioxidant, antimutagenic, anticarcinogenic, hypolipidemic, and cardioprotective activities (Chika and Bello, 2010). The antioxidant, antimicrobial, antidiarrheal, and anti-inflammatory properties of the plant are also well documented (Touré et al., 2011; Abdullahi et al., 2014). The process of drug discovery has always been challenging and arduous. It is time-consuming and financially overburdening in addition to having associated methodological complexities and limitations. In recent time, the advent of computer-aided drug discovery methods has revolutionized the process of drug discovery, design, and optimization (Pinzi and Rastelli, 2019; Vardhan and Sahoo, 2020). Molecular docking is one of the most frequently used methods in structure-based drug design (SBDD) that analyzes the conformation and orientation (pose) of molecules into the binding site of a macromolecular target. Search algorithms generate possible poses, which are then ranked by scoring functions with a substantial degree of accuracy (Ferreira et al., 2015; Toress et al., 2019). Docking was originally developed to help understand the mechanisms of molecular recognition between small and large molecules; however, the uses and applications of docking in drug discovery have increased tremendously. It enables researchers to identify novel compounds of therapeutic interest prior to in vitro or in vivo assays, predict ligand-target interactions at the molecular level, and delineate structure-activity relationships (SAR) without knowing a priori the chemical structure of other target modulators (Pinzi and Rastelli, 2019).
In recent studies, we have demonstrated the antioxidant and antidiabetic activity of the extracts of leaves of G. sylvestre and C. micranthum and the synergistic effect of the most active fractions of the two plants in vitro and in vivo, and also identified some putative, active compounds from the active fractions (Ibrahim et al., 2017, 2018). The present study aimed to conduct molecular docking and screening of ADME/drug-likeness properties of the identified potentially active candidate compounds from aqueous-methanol extracts of G. sylvestre and C. micranthum leaves by using an in silico pharmacoinformatics approach with a view to validate and prioritize promising compounds for further studies.
Materials and methods
Potential antidiabetic compounds
The analyzed compounds were previously isolated from the leaves of fresh G. sylvestre (GS) and C. micranthum (CM) collected from the Shira Local Government Area of Bauchi State in Nigeria. The detailed descriptions of the extraction, activity-guided isolation, and identification of the compounds have been reported in our previous study (Ibrahim et al., 2017, 2018). The compounds were identified by LC-MS analysis (UHPLC-ESI-Q-TOF-MS/MS) and NMR.
Ligand data
All the structural data of the identified compounds and acarbose were downloaded from PubChem or drawn with PubChem sketcher. Structure files were converted to the required format by using Open Babel.
Protein target data
The X-ray crystal structure data of human transferase (phosphorylated insulin receptor tyrosine kinase) (PDB ID: 1IR3) in peptide substrate and its analog; α-glucosidase (Halomonas sp. H11) (PDB ID: 3WY2) in complex with glucose and glycerol; and human pancreatic α-amylase (PDB ID: 4W93) in complex with montbtretin A were downloaded from the RCSB database (http:www.rcsb.org/pdb). The protein structure data were selected through literature analysis.
Molecular docking Ligand preparation
All ligand structures were optimized using Merck Molecular Force Field (MMFF) and the semi-empirical Austin Model (AM1) method, both of which are implemented in Spartan 14 software (version 14.1.4, wave function Inc., http://www.wavefun.com/). The fully optimized 3D structure without symmetry restrictions were saved as SD file through the file option on the Spartan 14 GUI.
Docking simulations
The preparation and minimization of all proteins were performed using tools and protocols in the Molsoft ICM-pro 3.8-3. The target protein structures were converted from PDB coordinates to ICM objects by using the object conversion protocol implemented in ICM-Pro. This process includes the addition of hydrogen atoms; assignment of secondary structures and energetically favorable protonation states to His, Asn, and Gln side chains, and of formal charges to the ligand in a complex with the receptor; and local minimization of polar hydrogen atoms using energy minimization protocols in ICM-Pro. The SD files of the ligands were then imported into ICM pro and used to dock the prepared receptors for targeting the active sites described in the literature (Hubbard, 1997; Brayer et al., 1995; Williams et al., 2012; Shen et al., 2015). The ligands were scored using the Biased Probability Monte Carlo (BPMC) procedure.
In silico and ADME and drug-likeness prediction
The in silico ADME screening and drug-likeness evaluation were performed using the free web tool SwissADME, which is developed by the Swiss Institute of Bioinformatics and freely available at www.swissadme.ch (Daina et al., 2017). The compounds with high-ranking binding energy scores were subjected to this part of the screening process. Simple physicochemical properties such as molecular weight (MW), molecular refractivity (MR), atom counts, and polar surface area (PSA) were computed. This was implemented by the OpenBabel, version 2.3.0 (O’Boyle et al., 2011). Drug-likeness candidature was implemented by Lipinski (2001), Ghose (1999), Veber (2002), Egan (2000), and Muegge (2001) rules of 5 (RO5) screening. The Abbot Bioavailability scores were computed to predict the probability of a compound to have at least 10% oral bioavailability by relying on total charge, TPSA, and violation of the Lipinski’s filter. Lipophilicity was implemented with iLOGP, XLOGP3, WLOGP, MLOGP, and SILICOS-IT models from which a consensus log Po/w was determined (Daina et al., 2017). The solubility (log S) of the selected ligands was implemented by three different models: ESOL (Delaney, 2004), Ali (Ali et al., 2012), and SILICOS-IT (Daina et al., 2017).
Results
Molecular docking
Table 1 lists the potential antidiabetic compound previously identified in the active fractions Gymnema sylvestre, Combretum micranthum extracts that was used for this study. Table 2 presents the docking scores/interaction for the identified compounds: A (2,2-dimethyl-3-[4-(acetyloxy)phenyl]-4-ethyl-2H-1-benzopyran-7-ol acetate), B ((3S)-6-hydroxy-3-isopropenyl-heptanoate), C (4-methyl-undecanoic acid), D (9,13-di-cis-retinoic acid), E (4-hydroxycinnamic acid), F ((–)-11-hydroxy-9,10-dihydrojasmonic acid), G (colnelenic acid), H (glyinflanin A), I (6,8a-seco-6,8a-deoxy-5-oxoavermectin “2a” aglycone), J (3-deshydroxysappanol trimethyl ether), and K (2,3,4,5-tetrahydroxy-cyclohexane ester glucoside) and the protein targets: α-amylase, α-glucosidase, and phosphorylated insulin receptor tyrosinase kinase. For pancreatic α-amylase, the docking score ranged from –2.746 to (–24.106) kcal/mol. The binding energies of compounds A, D, E, and H did not differ significantly from that of the reference standard acarbose (L) (–22.219 kcal/mol). For α-glucosidase, the docking score ranged from –1.696 to (–27.331) kcal/mol. Compounds E and H showed higher binding energy relative to the reference standard acarbose. Similarly, the docking score range of 0.450 to (–15.724) kcal/mol was observed for the phosphorylated insulin receptor tyrosine kinase. Compounds D, E, F, G, H, I, and J ranked higher than the reference standard acarbose in terms of the docking scores. Thus, compounds A, D, E, F, G, H, I and J showed potential to interact significantly with at least one of the three protein targets.
Table 1
Compound composition of subfraction 4 of fraction D [N-hexane : ethylacetate (20 : 80)] for G. sylvestre aqueous/methanolic leaf extract using LC/MS in the negative ion mode
Table 2
ICM docking scores (Kcal/mol) for compounds (ligands) from sub-fractions of extracts of G. sylvestre and C. micranthum
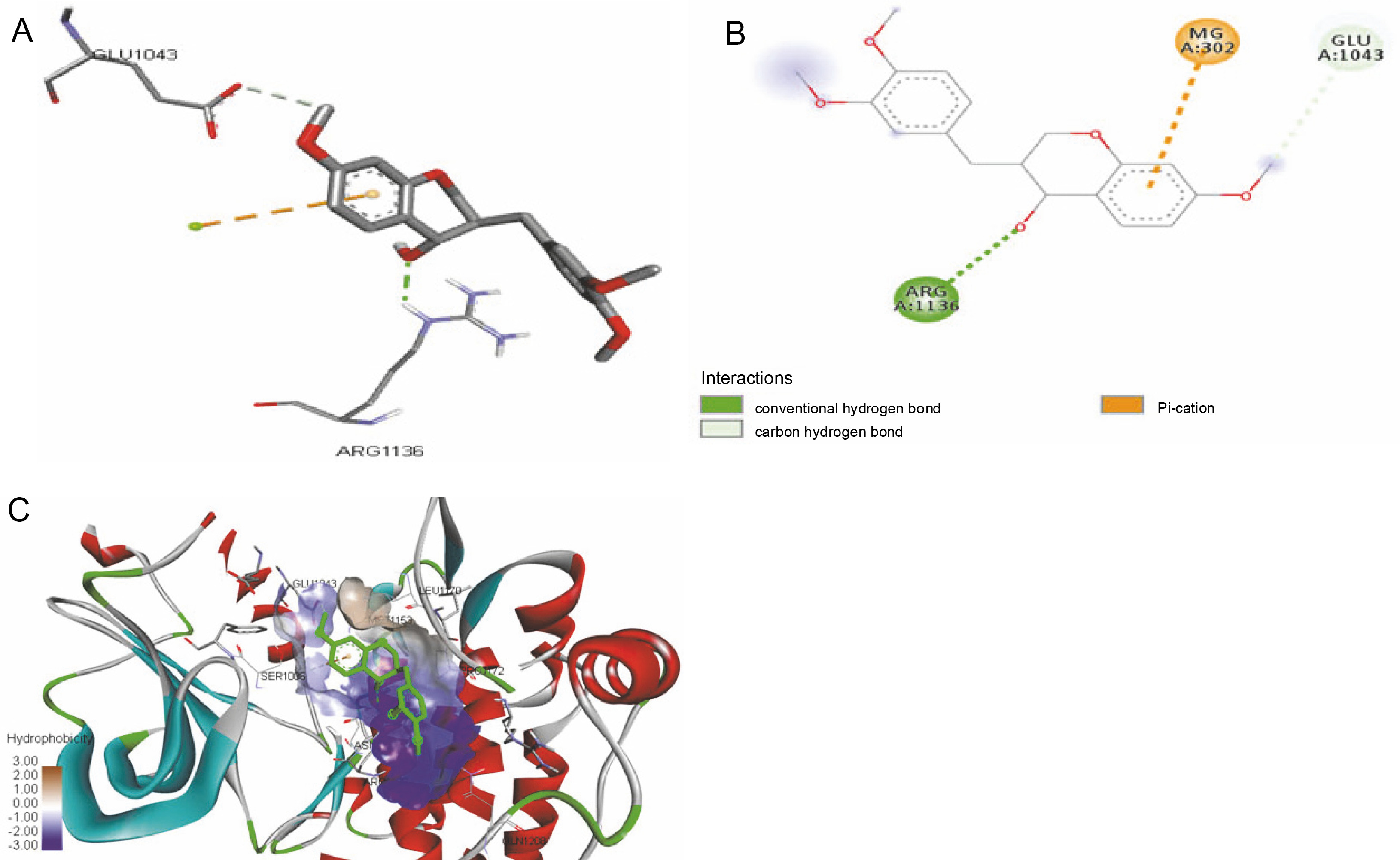
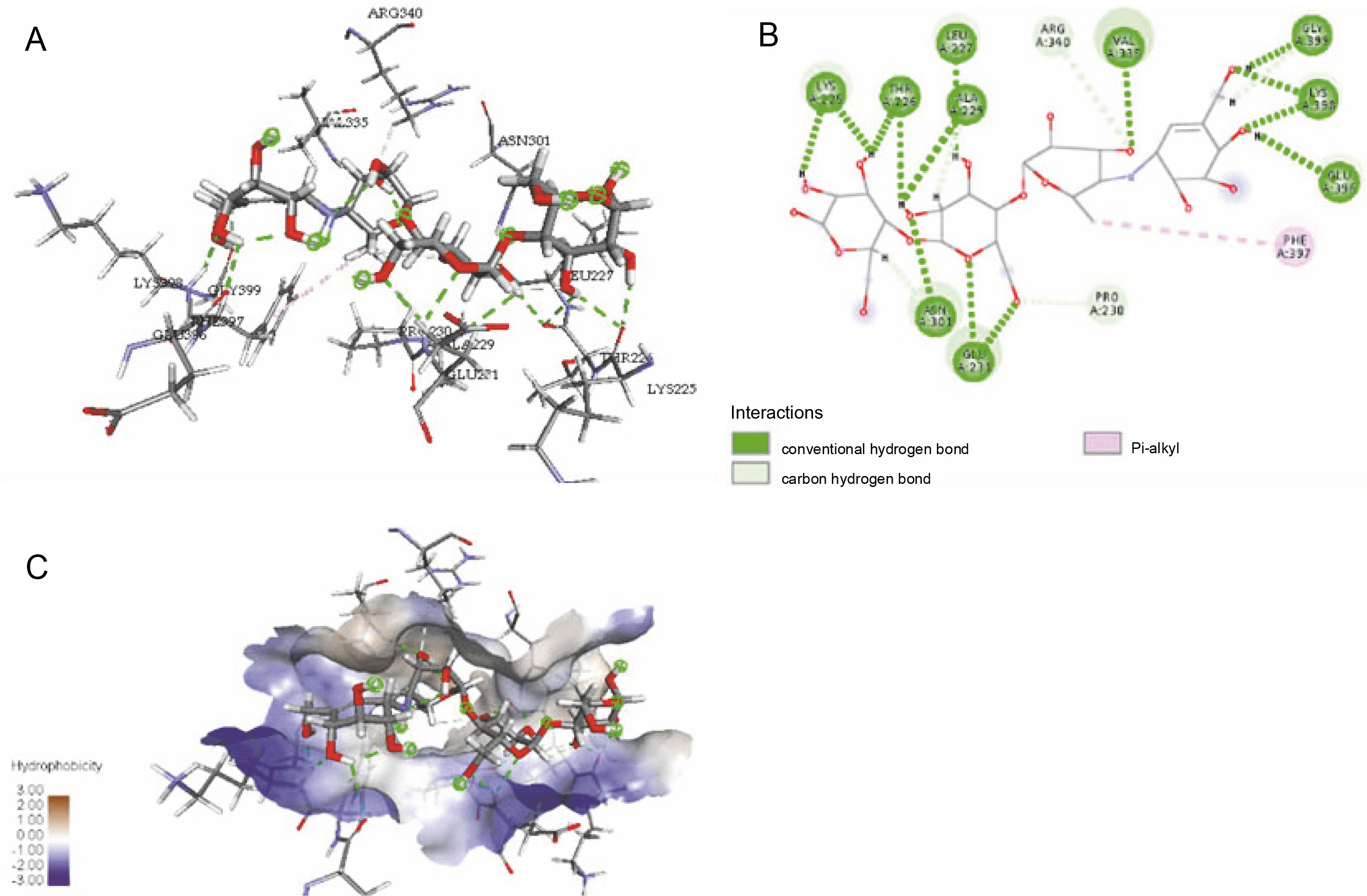
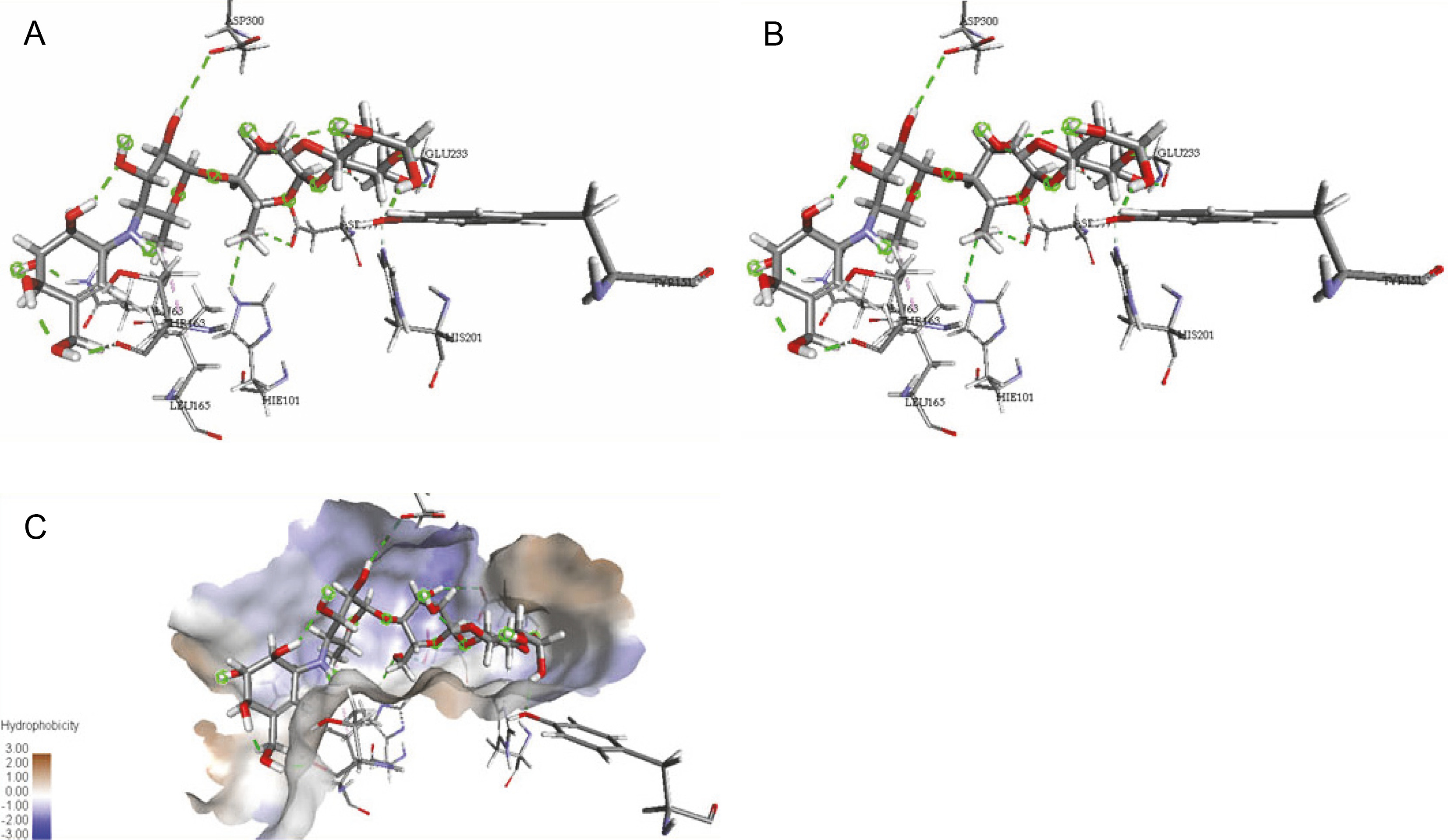
The ligand-protein interactions between the target proteins, the higher ranking compounds (in terms of binding energy), and acarbose (a standard drug) are presented in Figures 1–6. The predicted pose and interaction between 2,2-dimethyl-3-[4-(acetyloxy)phenyl]-4-ethyl-2H-1-benzopyran-7-ol and α-amylase (Fig. 1) show that the interactions were stabilized by hydrogen bonds formed by the amino acid residue Arg 389; van der Waals forces by residues Phe 315, Thr 377, Asp 375, Gln 390, and Lys 322; a pi-cation bond by Arg 343; a carbon-hydrogen bond involving Trp 316; a pi-pi stacked bond with Trp A 388; and some alkyl (pi-alkyl) bonds by residue Ala A 318. For glyinflanin A, α-glucosidase interactions were stabilized by four hydrogen bonds involving Asn 4, Trp 7, Gly 94, and His 459; van der Waals interactions involving the residues Trp 8, Asp 464, Lys 483, Leu 462, and Arg 456; and other bonds including a pi-anion with the residue Asp 48 and five alkyl (pi-alkyl) bonds formed by the amino acid residues Lys 96, Phe 463, Pro 460, and Arg 457 (Fig. 2). The interaction of (–)-11-hydroxy-9,10-dihydrojasmonic acid with phosphorylated insulin receptor tyrosine kinase (1IR3) was stabilized by amino acid residues Glu 1043, Arg 1155, Arg 1131, Arg 1039, and PTR 1163 forming hydrogen bonds (Fig. 3). The interaction of 3-deshydroxysappanol trimethyl ether with phosphorylated insulin receptor tyrosine kinase (1IR3) was stabilized by amino acid residues Arg 1136 (hydrogen bond), Glu 1043 (carbon-hydrogen bond), and a metal pi-cation bond with Mg 302 (Fig. 4). For the standard drug acarbose, the predicted binding pose with α-glucosidase (PDB ID: 3WY2) showed interactions stabilized by hydrogen bonds with Lys 225, Thr 226, Ala 229, Leu 227, Val 335, Gly 399, Lys 398, Glu 396, Glu 231, and Asn 301; carbon-hydrogen bond with Arg 340; and a pi-alkyl bond with Phe 397. Similarly, the standard drug also showed huge interactions with human pancreatic α-amylase (PDB ID: 4W93), which were stabilized by hydrogen bonds with Thr 163, Gln 63, Asp 300, His 101, Asp 197, Glu 233, and Tyr 151; carbonhydrogen bond with His 201; and a pi-alkyl bond with Leu 165. These interactions may largely account for the high docking scores recorded for these compounds and for their activity.
Fig. 1
ICM docking predicted pose for 2,2-dimethyl-3-[4-(acetyloxy)phenyl]-4-ethyl-2H-1-benzopyran-7-ol acetate in human pancreatic alpha-amylase (PDB ID: 4W93) showing: (Panel A and B) interactions with amino acid residues of target protein in 3D and 2D respectively; (Panel C) the compound docked in the binding pocket of the protein
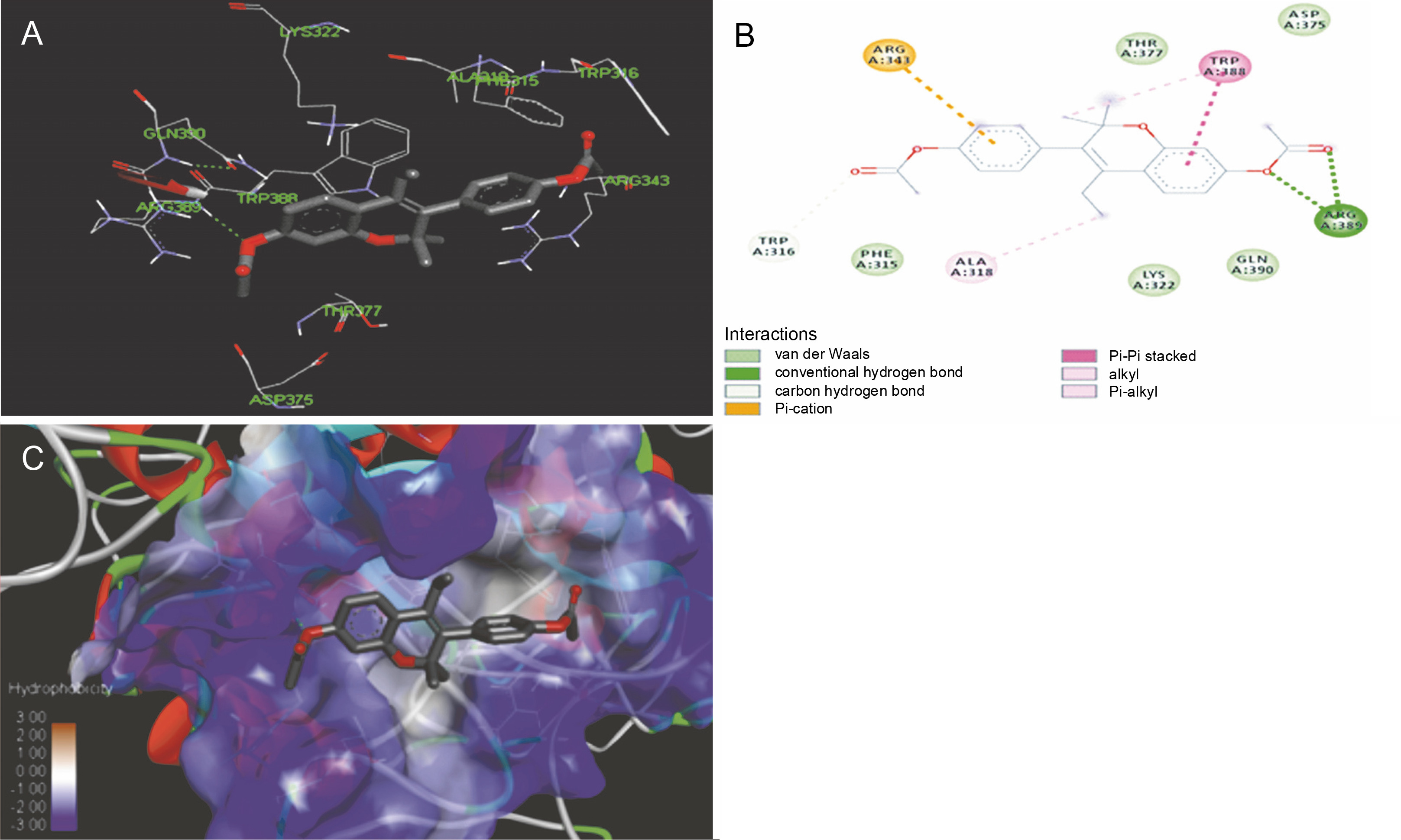
Fig. 2
ICM Docking predicted pose for glyinflanin A in alpha-glucosidase (PDB ID: 3WY2), showing: (Panel A and B) interactions with amino acid residues of target protein in 3D and 2D respectively; (Panel B) the compound docked in the binding pocket of the protein

Fig. 3
ICM docking predicted pose for (–)-11-hydroxy-9,10-dihydrojasmonic acid in phosphorylated insulin receptor tyrosine kinase (PDB ID: 1IR3) showing: (Panel A and B) interactions with amino acid residues of target protein in 3D and 2D respectively; (Panel C) the compound docked in the binding pocket of the protein
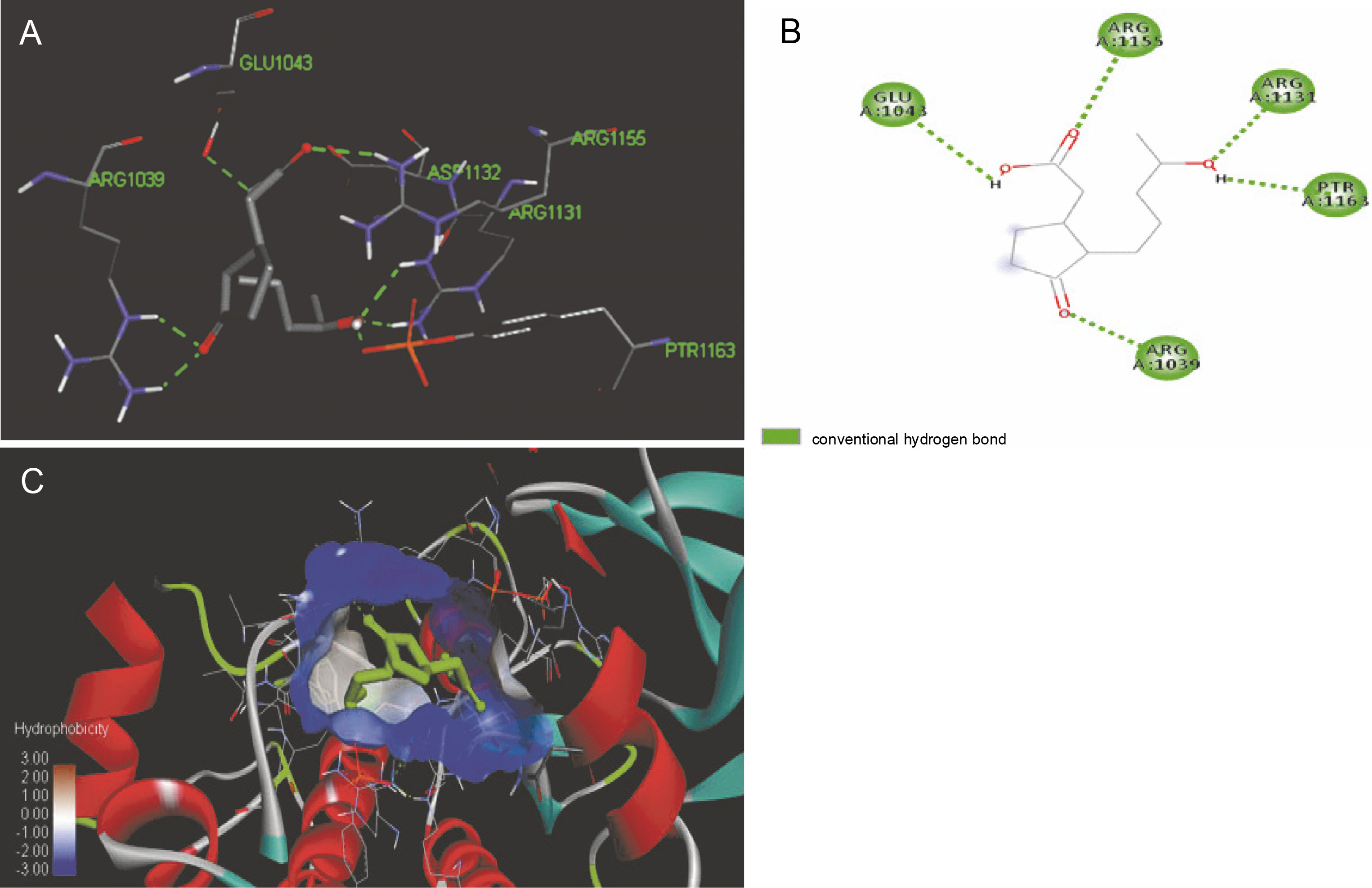
Fig. 4
ICM docking predicted pose for 3-deshydroxysappanol trimethyl ether in phosphorylated insulin receptor tyrosine kinase (PDB ID: 1IR3) showing: (Panel A and B) interactions with amino acid residues of target protein in 3D and 2D respectively; (Panel C) the compound docked in the binding pocket of the protein
Physicochemical properties of the selected compounds
The basic physicochemical properties of the promising compounds from the docking result are presented in Tables 3, 4, and 5. The molecular weights of the selected compounds ranged from 164.16 to 645.6 g/mol. Only compound L (acarbose) had a molecular weight higher than 500 g/mol. The computed consensus LogP value (lipophilicity) for the selected compounds ranged from 1.26 to 5.01, except for acarbose that showed a significantly lower and negative lipophilicity (–6.22 ). The computed water solubility index showed that all the selected compounds were soluble in water but differed in their degree of solubility – acarbose showed a relatively high solubility.
Table 3
Basic physicochemical properties and computational descriptors of the screened compounds
Table 4
Lipophicility of the screened compounds
Table 5
Predicted water solubility of the screened compounds
ADME parameters
The computed ADMET properties (gastrointestinal (GI) absorption; blood-brain barrier (BBB) permeation; inhibition of the cytochrome P450 system; permeability glycoprotein (P-gp) substrate) of these compounds are presented in Table 6. According to the results, all the compounds showed high gastrointestinal absorption, except for compounds I and L. BBB permeation potential was predicted for compounds A, E, G, D, and J. Compounds I and L showed potential to be a substrate of P-gp. The potential to inhibit cytochrome P450 (CYP) isoforms was observed for compounds A (for 3 isoforms); G , H, D, and J (for 2 isoforms); and I (for one isoform). Compounds E, F, and L were predicted to show no inhibitory activity against any of the CYP isoforms.
Table 6
Predicted pharmacokinetics (ADME) parameters of the screened compounds
Drug-likeness and medicinal chemistry parameters of the selected compounds
The computed drug-likeness, medicinal chemistry, and lead-likeness properties of the selected compounds are presented in Table 7. According to the results, compounds A, F, and J showed no violation of all five filters (Lipinski, Ghose, Veber, Egan, and Muegge) used. The other compounds showed violation of at least one of the drug-likeness filters. The computed Abbot Bioavailability score for all the compounds placed them within the 55–56% probability class, except for compound L (0.17). All the compounds showed at least one lead-likeness violation and BRENK alert.
Table 7
Predicted drug-likeness, medicinal chemistry and lead-likeness pharmacokinetics parameters of the screened compounds
Discussion
Molecular docking is an in silico modeling technique used to study the interaction between protein targets and small ligands (compounds) (Iheagwam et al., 2019). The docking score or binding energy derived from the docking is considered to be a function of the binding affinity of the ligand to the protein target. In the present study, the docking of potentially active compounds (identified from active subfractions of G. sylvestre and C. micranthum extracts) (Ibrahim et al., 2017, 2018) against the target proteins human pancreatic α-amylase, α-glucosidase, and phosphorylated insulin receptor tyrosine kinase was performed. The compounds A (2,2-dimethyl-3-[4-(acetyloxy)phenyl]-4-ethyl-2H-1-benzopyran-7-ol acetate), D (9,13-di-cis-retinoic acid), E (4-hydroxycinnamic acid), F ((–)-11-hydroxy-9,10-dihydrojasmonic acid), G (colnelenic acid), H (glyinflanin A), I (6,8a-seco-6,8a-deoxy-5-oxoavermectin “2a” aglycone), and J (3-deshydroxysappanol trimethyl ether) showed a relatively high potential to bind and interact with at least one of the three target proteins screened. The observed docking scores (binding energy) were comparable to that of the reference drug L (acarbose). Overall, the docking interaction results showed that most of the compounds with high binding affinity (in terms of docking score) also exhibited many interactions with the active site residues of the target protein. Some compounds such as (–)-11-hydroxy-9,10-dihydrojasmonic acid and 3-deshydroxysappanol trimethyl ether showed hydrogen bond interaction with the active site catalytic residues (Glu 1043, PTR 1163, and Arg 1136) of phosphorylated insulin receptor tyrosine kinase (Hubbard, 1997) and acarbose, and also with the catalytic residues (Glu 233, Asp 300, and Asp 197) of α-amylase. This is expected to significantly affect the activity of the target proteins and result in inhibitory modulation of their activity. On the other hand, the interactions between 2,2-dimethyl-3-[4-(acetyloxy)phenyl]-4-ethyl-2H-1-benzopyran-7-ol acetate/α-amylase and glyinflanin A/α-glucosidase were not directly linked to active site residues, but rather to residues outside the active site cleft. However, such interaction is also capable of affecting the activity of the target proteins by disordering the polypeptides that constitute the active site as observed by William et al. (2012) in their study with myricetin and ethyl caffeate. Thus, this molecular docking study corroborates the inhibition of the enzymes α-glucosidase and α-amylase reported earlier by Ibrahim et al. (2017) and further supports the postprandial glucose lowering ability of the plant extract. This is a strong indication that these compounds could have contributed significantly to the observed hypoglycemic activity of the extracts of the two plants: they may act synergistically to produce the observed activities (E showed high binding energy to all the three target proteins, F to two, H to two, and J to one). This could be attributed to the structural similarity between the conventional substrates or ligands of the target proteins and the compounds isolated from the plants.
For a potential compound to be considered as a candidate drug for eventual use, it must possess apt or optimal pharmacokinetics and safety characteristics in addition to the desired biological activity (Hu et al., 2018). Hence, we evaluated the selected promising compounds from the docking simulation to determine apt pharmaco-kinetics, drug-likeness, and optimal medicinal chemistry characteristics by using SwissADME in silico suite. Lipophilicity of a drug is a measure of how well the drug dissolves in lipids or nonpolar solvents. This property has a huge effect on the overall ADMET property of the drug. It plays a major role in the absorption of drugs across cell membranes (Arnott and Planey, 2012). According to most filters (rule of 5) for drug-likeness, a lipophilicity range of 0 to 5 is usually considered optimal for drug design (Waring et al., 2010; Lipinski et al., 2001). In the present study, compounds A (2,2-dimethyl-3-[4-(acetyloxy)phenyl]-4-ethyl-2H-1-benzopyran-7-ol acetate), E (4-hydroxycinnamic acid), F ((-)-11-hydroxy-9,10-dihydrojasmonic acid), G (colnelenic acid), H (glyinflanin A), I (6,8a-seco-6,8a-deoxy-5-oxoavermectin “2a” aglycone), J (3-deshydroxysappanol trimethyl ether), and L (acarbose) showed optimal lipophilicity. This implies that these compounds will be well absorbed through the membranes into the systemic circulation to achieve high bioavailability. Solubility, on the other hand, is a physicochemical property of drugs that affects absorption, distribution, and formulation (Daina et al., 2017). To facilitate their absorption, drugs are expected to be present in the form of aqueous solution at the site of absorption (Savjani et al., 2012). In the present study, all the screened compounds showed relatively moderate solubility, an indication that these compounds (with optimal lipophilicity and moderate solubility) can achieve good bioavailability when administered orally. However, compound L (acarbose) showed an extremely high solubility and low lipophilicity. This may result in poor absorption into the bloodstream from the gastrointestinal tract (GIT). However, the consequence of this may not be grave, as the site of action of drug L is the GIT, where it is expected to interphase with and inhibit amylase and glucosidase enzymes (Rosak and Mertes, 2014). The other compounds/drugs that may have alternative target sites (insulin-like, metformin-like, glibenclamide-like acting compounds) in addition to the GIT will require relatively improved lipophilicity and solubility to enhance the absorption of the drug beyond the GIT and also achieve effective concentration at the target sites.
ADME properties of drugs determine their eventual fate in the body (Shin et al., 2016). Drugs for oral administration should be well absorbed in the GIT for optimal pharmacokinetics. The BBB is very important to limit the penetration of drugs into the central nervous system (CNS) (Abbott, 2002). In the present study, it was observed that all the compounds have potential to be well absorbed into the systemic circulation in the GIT (except for compound I (6,8a-seco-6,8a-deoxy-5-oxoavermectin “2a” aglycone) and compound L (acarbose)) and permeate the BBB. This may be a cause of concern for possible CNS-related side effects. However, because the computational method does not quantify the extent of permeation, the degree may not be so significant where it induces marked toxic effect on the CNS.
P-gp usually acts as an efflux transporter that pumps xenobiotics or drugs back into the GIT lumen and consequently reduces plasma and tissue concentrations of the drug (Kim, 2002; Finch and Pillians, 2014). In the present study, all the compounds were predicted to be nonsubstrate of P-gp, except for compounds I (6,8a-seco-6,8a-deoxy-5-oxoavermectin “2a” aglycone) and L (acarbose). This may not be a serious concern for the standard drug acarbose, whose action of targeting α-amylase and α-glucosidase activity occurs in the GIT. However, for compounds such as I, whose target (insulin receptor-associated protein) is outside the lumen of the gut, this may affect their bioavailability at the target site, as the absorbed drug molecules may be pumped back into P-pg.
The knowledge of interactions between compounds and the cytochrome P450 (CYP) system is very essential to characterize the pharmacokinetics of candidate drugs as these interactions are necessary for the transformation and elimination of drugs from the system (Daina et al., 2017). Inhibition of isoforms of this enzyme system by drugs could result in poor elimination, resulting in drug-induced toxicity. It is therefore important that a candidate drug should have limited inhibitory activity against these enzyme isoforms. The present study revealed that compounds E (4-hydroxycinnamic acid), F ((–)-11-hydroxy-9,10-dihydrojasmonic acid), and L (acarbose) showed no potential to inhibit any of the five P450 isoforms. This indicates that these compounds would be well metabolized in the liver and eliminated easily from the organism.
The overall drug-likeness property assesses quantitatively how much of the physicochemical and structural characteristics of the compounds comply or are consistent with the majority of well-known drugs. This was predicted using five computational “rule of 5” filters – Lipinski (Pfizer), Ghose (Amgen), Verber (GSK), Egan (Pharmacia), and Muegge (Bayer) – to allow for consensus in the prediction (Daina et al., 2017). The Lipinski’s rule is the first-ever implemented “rule of 5” for drug-likeness, and the result of this study showed that six of the compounds (A, E, F, G, H, and J) screened showed compliance with the Lipinski’s rule. On using multiple filters (to increase the precision of the prediction), compounds A (2,2-dimethyl-3-[4-(acetyloxy) phenyl]-4-ethyl-2H-1-benzopyran-7-ol acetate), F ((–)-11-hydroxy-9,10-dihydrojasmonic acid), and J (3-deshydroxysappanol trimethyl ether) showed no violation of any rules for all 5 filters. A violation of Muegge was observed for compound E and of Ghose and Muegge for compound H. All the other compounds showed violation of more than 3 of the 5 filters. This finding suggests that A (2,2-dimethyl-3-[4-(acetyloxy)phenyl]-4-ethyl-2H-1-benzopyran-7-ol acetate), E (4-hydroxycinnamic acid), F ((–)-11-hydroxy-9,10-dihydrojasmonic acid), H (glyinflanin A), and J (3-deshydroxysappanol trimethyl ether) compounds have better drug-like characteristics relative to the other compounds.
The compounds were also screened for BRENK and PAIN alerts. None of the studied compound in this study showed any PAIN alert. This indicates the absence of any structurally promiscuous moiety (otherwise known as frequent litters). These structural fragments or components are known to produce false-positive responses in in silico assays regardless of the protein target being studied (Baell and Holloway, 2010). Such alerts should be treated with caution. BRENK alerts, on the other hand, depict fragments of compounds that could be putatively toxic, chemically reactive, and metabolically unstable. This study revealed that all the compounds showed at least one BRENK alert, except F ((–)-11-hydroxy-9,10-dihydrojasmonic acid) and J (3-deshydroxysappanol trimethyl ether). However, E (4-hydroxycinnamic acid), H (glyinflanin A), I (6,8a-seco-6,8a-deoxy-5-oxoavermectin “2a” aglycone), and L (acarbose) showed only one BRENK alert each. This may not be a serious concern as it depends on the nature of the structural moiety producing the alert. Nonetheless, this aspect should be factored into prioritization of compounds for drug-likeness qualities. Overall, compounds E, F, H, and J seem to have the most promising characteristics in terms of binding interactions with target proteins and ADMET and drug-likeness properties. To the best of our knowledge, these compounds have not been reported elsewhere as antidiabetic agents.
Conclusions
The present study investigated potential antidiabetic compounds identified in a previous report from active subfractions of G. sylvestre and C. micranthum extracts by using in silico techniques. The results validated the antidiabetic potential of the extract, with some of the identified compounds showing strong binding affinity with target proteins relative to the reference drug (acarbose). Furthermore, the drug-likeness screening of the promising compounds identified (prioritized) four druglike and lead-like candidates: E (4-hydroxycinnamic acid), F ((–)-11-hydroxy-9,10-dihydrojasmonic acid), H (glyinflanin A), and J (3-deshydroxysappanol trimethyl ether) from the set. These compounds can now be further evaluated individually for antidiabetic activity, definite mechanism of action, and optimization in further studies.