









Introduction
Cardiac tumors include metastases and primary tumors, with metastases being more common [1]. Primary cardiac tumors are benign in the majority of cases, with atrial myxoma the most common type in adults and rhabdomyosarcoma the most common in children [2, 3]. Rhabdomyosarcoma, however, can be seen at any age and can present with symptomatology from the cardiovascular and the respiratory system [3]. Primary rhabdomyosarcomas, although rare, have been reported in the literature, with left atrial (LA) the most common location [3, 4]. Rhabdomyosarcomas involving both atria represent an extremely rare diagnosis with only 2 cases, to our knowledge, reported in the literature to date [5, 6].
We therefore present a rare case of a 63-year-old woman diagnosed with multiple embryonal rhabdomyosarcomas of both atria that required surgical excision. To our knowledge, this is the third case of biatrial rhabdomyosarcoma.
Case report
A 63-year-old female patient presented with progressive exertional and nocturnal palpitations, cough and dyspnea. She was on sinus rhythm, and she did not have a history of any medications. Early diastolic and systolic murmurs in the mitral valve auscultation area were noted.
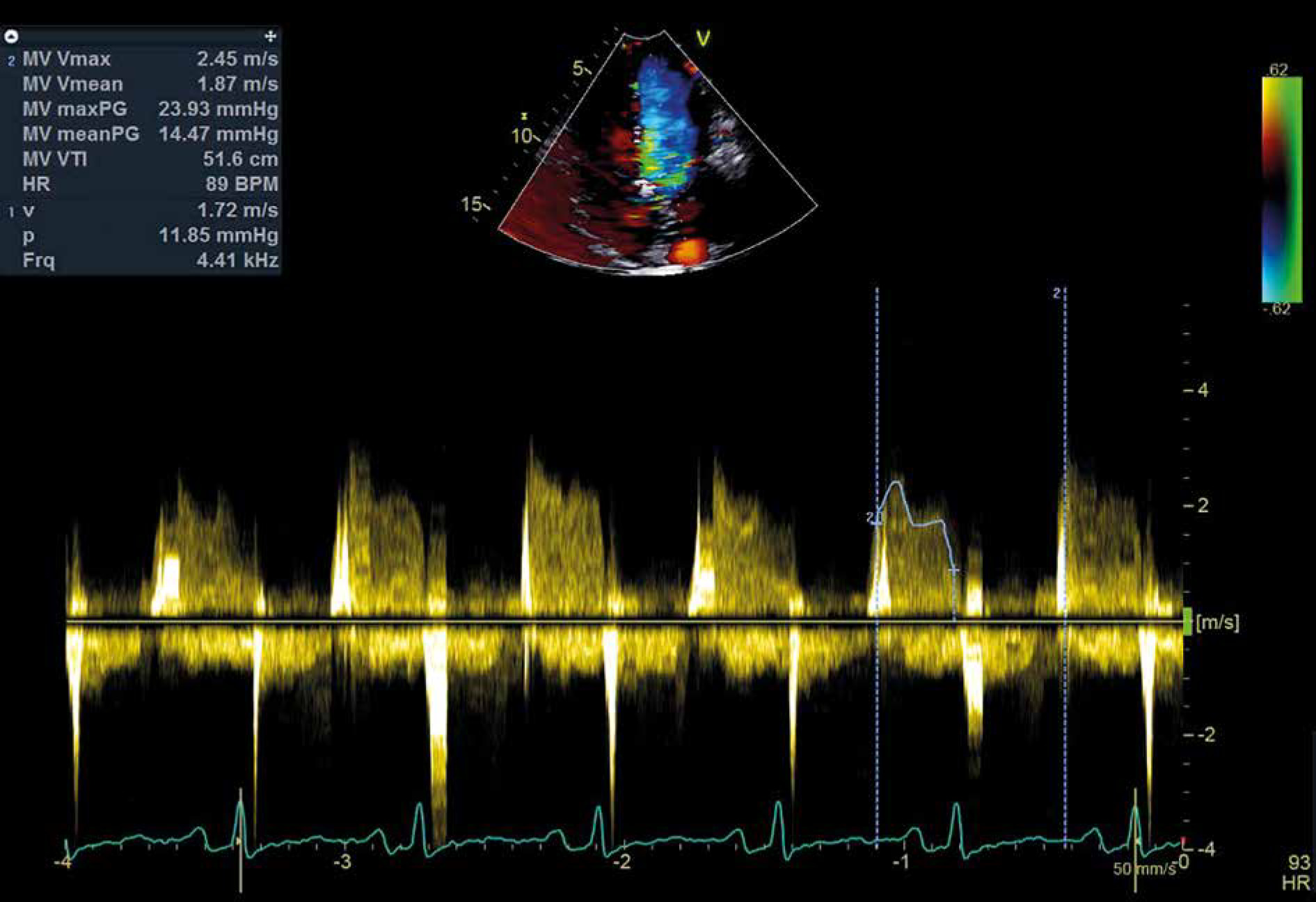
A transthoracic echocardiogram (TTE) was performed, which revealed a large, solid, mobile, irregular cardiac mass. The mass was arising from the posterior LA wall and located within the LA, while also protruding into the left ventricle (LV). The mitral valve pressure gradient was calculated at 15 mm Hg, which was consistent with moderate mitral stenosis (Fig. 1). The right atrium (RA) was also enlarged, and the pulmonary pressure was extremely elevated (70 mm Hg). No further valve abnormalities or LV hypokinetic areas were detected, and the cardiac performance was normal with ejection fraction (EF) of 60% (EF ≈ 60%).
Fig. 1
Transthoracic echocardiogram revealed a mitral valve pressure gradient of 15 mm Hg, consistent with moderate mitral stenosis
The presumptive initial diagnosis of cardiac tumor, such as atrial myxoma, was made and operative resection was therefore recommended. The patient was staged with a combination of imaging modalities that included computed tomography (CT) scans and bone scintigrams, and PET-CT that did not reveal any signs of disease spread to adjacent organs, or distant metastases. A multidisciplinary team meeting, including cardiologists, cardiac surgeons and oncologists, decided that the optimal course of treatment was to attempt a radical surgical resection that would be followed by adjuvant chemotherapy and radiation treatments.
The operation was performed under moderate hypothermia, through a median sternotomy approach. The ascending aorta and both caval veins were cannulated, cardiopulmonary bypass was conducted, and the operation was performed. Myocardial protection was achieved with antegrade cold blood cardioplegia. Intraoperatively, however, 4 distinct masses were revealed in both atrial cavities; 3 were located in the LA and 1 in RA (Fig. 2). The larger tumor consisted of two lobulated yellow components and was attached with a broad base to the posterior LA wall. The left atrial mass was approached through the interatrial septum (transseptal approach) and all 4 tumors were removed en bloc and sent for biopsy (Fig. 3). Care was taken not to manipulate the tumor before the cross-clamp had been applied on the aorta. The use of cardiotomy vents was minimal to avoid vacuuming of the tumor fragments. Right atriotomy and septotomy (excision of the fossa ovalis) were performed; a pannus-like neoplastic tissue involving the pulmonary vein orifices was also excised. No tumor infiltration of the atrial or ventricle walls was detected. The mitral valve was also normal with no evidence of tumor invasion.
Fig. 2
Four primary rhabdomyosarcomas were excised; three were located in the left atrium and one into the right atrium
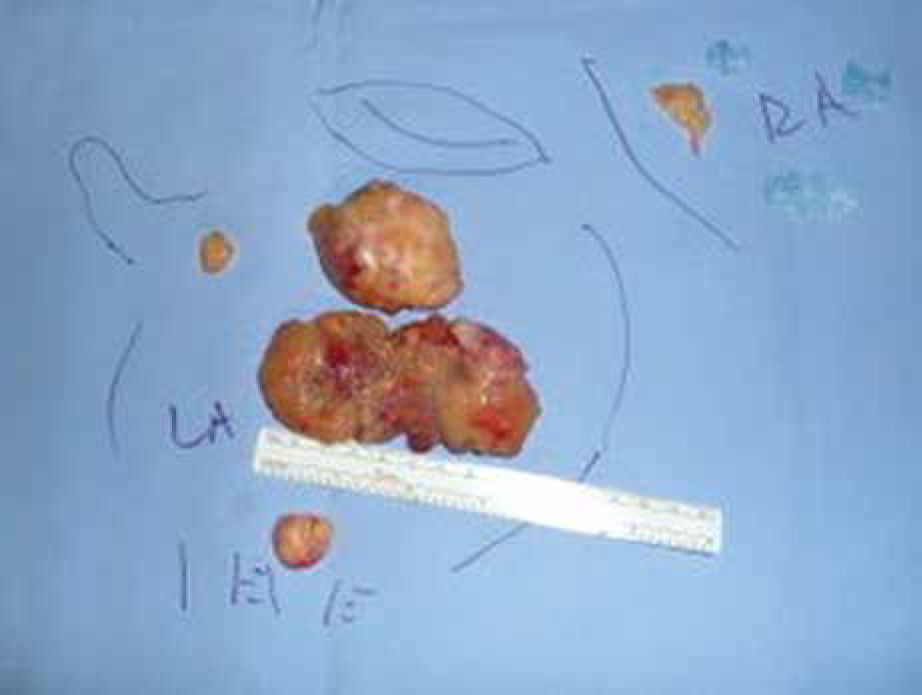
Fig. 3
All four rhabdomyosarcomas were excised en bloc through a transeptal approach and were sent for biopsy

Initial biopsy from intraoperative specimens confirmed the diagnosis of embryonal rhabdomyosarcoma on all 4 masses and that tumor margins were tumor-free. The final pathologic report characterized the tumor as an atrial rhabdomyosarcoma with botryoid features with an immunophenotype of Vim positive, SMA negative, CD56 positive, CD99 negative CD31 negative, CD68 positive and a Ki67 index of 80%. The pathological report was conclusive on the histological characterization, with no differential diagnoses being considered by two expert pathologists.
The postoperative course was uneventful and the patient was discharged on the 5th postoperative day. Postoperative cardiac functional tests revealed an EF of 60%, consistent with the preoperative value, and no mitral valve dysfunction.
After discharge, the patient was referred to the oncological team for further treatment, according to the initial treatment plan. The patient received chemotherapy and radiotherapy, but was subsequently lost to follow-up after 6 months. The oncological team informed us of her passing away 14 months after the initial diagnosis.
Discussion
Cardiac tumors comprise a heterogenous group arising either from metastases or primary cardiac tumors [1–3]. Metastases are more common, with prevalence of 7.1% in cancer patients and 2.3% in the general population [1], while the frequency of primary cardiac tumors is in the range 1.38–30 cases/100 000 population [2]. Primary malignant cardiac tumors are even rarer (20% of the primary cardiac tumors), of which rhabdomyosarcoma is the second most common sarcoma [3].
To date, almost 70 cases of cardiac rhabdomyosarcomas have been reported in the literature, with more than half of them (55%) located in the LA, followed by the LV, right ventricle (RV) and RA [3, 4]. However, biatrial rhabdomyosarcomas are extremely rare, with only 2 cases reported, to our knowledge, in the extant literature [5, 6]. Therefore, our case report is of the highest importance, since we present the case of a patient with extensive organ involvement in an already rare condition as a primary malignant intracardiac tumor. Due to the scarcity of large cohort reports on the condition, management decisions are extremely difficult, especially if one considers the biatrial involvement of the specific patient. Our report demonstrates that surgical treatment of large cardiac rhabdomyosarcomas with involvement of more than one cardiac chamber is a technically demanding, yet feasible management strategy that can lead to symptom resolution, discharge with normal cardiac function and seemingly non-inferior survival compared to rates reported in the literature for our patient. Although ours is a case report, the extremely low number of similar reported cases makes ours a valid treatment proposition, and a proof of concept, that radical surgery is a viable option in large, biatrial cardiac tumors, especially in patients in whom no evidence of metastatic disease is found at presentation.
The symptoms can vary and depend on the location, the size and the number of the masses [3]. Tumors arising from the RA or RV can present with symptoms of right heart failure (peripheral edema, ascites), while tumors of the LA or LV present with symptoms of left heart failure (dyspnea, easy fatiguability) [3]. If the tumor blocks the inflow or outflow of the mitral valve, it can present with arrythmias and symptomatology similar to left heart failure, as in our case. Cardiac tamponade should be excluded, as it needs emergent management. Patients can also present with syncope, embolic events, supraventricular or ventricular tachycardia, heart block and even cardiac death [7].
Imaging modalities available include, among others, TTE, CT and magnetic resonance (MR). Transthoracic echocardiogram is the initial and most commonly used method to diagnose the presence of intracardiac tumors and depicts well tumors located in the left atrium or near the mitral valve. Transthoracic echocardiogram is readily available even for the Emergency Department staff [8]; however, it cannot differentiate between benign and malignant mass [9, 10]. Computed tomography and MR provide better results in terms of mass visualization and tissue characterization compared to TTE, but they both have disadvantages. With CT, patients are exposed to ionized radiation and possible nephrotoxic iodinated contrast, and MR is not always readily available [3, 9, 10].
As imaging cannot confirm a distinct diagnosis, most masses are given an initial presumptive diagnosis of myxoma and proceed to excision and biopsy, as in our case. Both benign and malignant tumors may have similar symptomatology, rendering the correct diagnosis challenging [3]. Biopsy, either intraoperatively or after tumor excision, confirms the histological type of the mass [7, 9].
Rhabdomyosarcomas have a very poor prognosis with median survival less than a year [11]. Although there is no clear consensus on their optimal management, a multidisciplinary approach with surgical resection combined with chemotherapy or radiotherapy may benefit the patient [12]. More specifically, a combination of surgical resection with tumor-free margins and neoadjuvant chemotherapy provides almost triple survival compared to surgery or chemotherapy alone (36.5 months vs. 14.1 months) [12, 13]. Surgical resection alone did not provide optimal survival outcomes, with most experts agreeing that administration of adjuvant chemotherapy/radiotherapy is the optimal course of treatment for cardiac sarcomas. Another study by the French sarcoma group, enrolling one of the largest cardiac sarcoma patient cohorts to date, also confirms the importance of complete surgical resection with clear margins. Patients with satisfactory resection margins achieved a median overall survival of 38.8 months, compared to 18.2 for patients with infiltrated resection margins and 11.2 months in patients who were not treated surgically [14]. Chemotherapy and radiotherapy were both found to be associated with better progression-free survival rates when added to surgical resection, but chemotherapy was not found to contribute significantly to the overall survival of surgically treated patients [12, 14, 15]. Primary biatrial rhabdomyosarcomas have been rarely reported and there are no guidelines on how to approach them. Uchida et al. opted to surgically resect the tumor, but recurrence ensued and although the patient received molecular-targeted salvage chemotherapy, the patient died 13 months after surgical resection [5]. Tavil et al. opted to manage their patient with palliative chemotherapy only, as he was unstable and had metastasis already [6].
Conclusions
Primary biatrial rhabdomyosarcoma is an extremely rare diagnosis that can present with symptomatology based on the location, size and the number of masses. There is no consensus on how to manage them due to the scarcity of cases, but they are managed as single rhabdomyosarcomas. The majority require surgical excision, with subsequent chemotherapy or radiotherapy. Prognosis is very poor, with the majority of the patients not surviving longer than one year.