

Introduction
Primary combined immunodeficiency (CID) is a rare and complex group of genetic disorders, belonging to a significant category within CID. The main characteristic is the simultaneous impairment of T lymphocytes and B lymphocytes in patients, resulting in the dysfunction of both humoral and cellular immune systems [1]. While affecting only a small population globally, this disease has been a focal point of immunological and genetic research due to the severe threat it poses to patients’ lives. The pathogenesis of CID is rooted in genetic variations, often arising from mutations in crucial genes involved in the development and function of the immune system. These genetic mutations can be autosomal recessive, auto- somal dominant, or X-linked recessive, leading to impairments in critical processes of immune cell generation, differentiation, or signaling pathways [2]. Among these, X-linked severe combined immunodeficiency (X-SCID) is a well-known type caused by mutations in the IL2RG gene, which encodes the interleukin 2 receptor gamma chain crucial for the development of T cells and natural killer cells. This disease affects male infants, rendering them nearly defenseless against common pathogens such as bacteria, viruses, and fungi [3-5]. Individuals with X-SCID often experience recurrent severe infections due to this deficiency, which, if untreated, can lead to early mortality [4-6]. Apart from the classical clinical manifestations, patients may also exhibit delayed growth and severe malnutrition, further complicating the disease and its treatment [7, 8].
The IL2RG gene is located on the X chromosome and encodes the gamma chain, a component of various cell surface receptors crucial for the growth, differentiation, and survival of multiple immune cells [9, 10]. The gamma chain interacts with various cytokines (such as IL-2, IL-4, IL-7, IL-9, and IL-15) to regulate immune cell activity, including T cells and natural killer (NK) cells. In X-SCID patients, mutations in the IL2RG gene inhibit these critical immune response pathways, leading to a significant reduction in the number or impaired function of T cells and NK cells, resulting in a severe immune deficiency [7, 11, 12].
Despite significant advances in the diagnostic methods for CID and X-SCID, including the utilization of newborn screening and advanced molecular genetic technologies, substantial challenges still persist [13]. On one hand, CID patients with mild symptoms during infancy may not exhibit all typical signs or may present with mild symptoms, potentially leading to diagnostic delays [1]. On the other hand, the diagnostic process necessitates a comprehensive evaluation of clinical manifestations, immunological function tests, and genetic analyses, requiring medical teams with high levels of expertise and experience. Moreover, the treatment of more severe forms of CID, such as X-SCID, typically involves bone marrow transplantation or gene therapy, underscoring the critical importance of early diagnosis in improving patient survival rates and quality of life [6, 14]. Presently, research on X-SCID has yielded certain achievements internationally [15], yet the full elucidation of novel IL2RG gene mutations and their clinical distinction in CID and X-SCID remains incomplete. These studies hold significant implications for understanding the genetic heterogeneity of CID and X-SCID, refining diagnostic methodologies, and developing novel treatment strategies. Particularly, the identification of new mutations not only aids in uncovering the genetic basis of diseases but also potentially provides researchers with novel molecular targets, advancing the development of targeted therapeutic approaches [14, 16].
This study reports a case of CID in a 10-year-old male child who was initially misdiagnosed due to bronchiectasis. Further analysis revealed a heterozygous missense mutation c.420A>T(p.R140S) in the IL2RG gene. The aim of this study is to enhance healthcare professionals’ awareness of CID by thoroughly analyzing this specific case, especially when patients exhibit atypical symptoms or when routine genetic analyses fail to diagnose known pathogenic variations. Additionally, this study seeks to expand knowledge of the genetic spectrum of CID both domestically and internationally through the exploration of new genetic variations, offering fresh insights for early clinical diagnosis and treatment of CID. Through long-term follow-up and comprehensive management, this study also emphasizes the crucial role of genetic counseling in children and their families, particularly in coping with ongoing challenges such as sports activities, rhinitis, and sinusitis. Ultimately, this study delivers valuable information and novel directions to enhance the diagnosis, treatment, and management of CID patients.
Material and methods
Genetic analysis
DNA was extracted from the peripheral blood samples of participants using standardized methods for lymphocyte subpopulation isolation and purification. High-throughput sequencing of the extracted DNA was then performed using whole-exome sequencing (WES) technology on the Illumina HiSeq 1500 system, in conjunction with the Agilent SureSelect Clinical Research Capture Exome kit, aimed at capturing and sequencing the entire exome region. The data processing included quality control, sequence alignment, variant detection, and annotation steps to identify genetic variants potentially related to the disease phenotype. All identified variants were subjected to in-depth analysis using bioinformatics tools and databases to assess their potential functional impact and pathogenicity. To validate candidate variants and screen for mutations among family members, polymerase chain reaction (PCR) was employed to amplify target DNA fragments, followed by capillary electrophoresis on the ABI-3730XL DNA analyzer, and sequencing of the amplified fragments using the BigDye Terminator cycle sequencing kit. Additionally, to accurately assess the presence of variants across different lymphocyte subpopulations, targeted screening of specific lymphocyte subpopulations was conducted using PCR amplification and capillary electrophoresis techniques.
Immunological assay basics
Mononuclear cells were isolated from blood samples using Ficoll-Paque PLUS (GE Healthcare) density gradient centrifugation. The separated PBMCs were then incubated on ice with fluorescently conjugated antibodies for 30 minutes to label specific cell surface markers. The antibodies used included fluorescent-labeled anti-human CD4 (BioLegend), CD3 (ImmunoTools), and CD8 (Miltenyi Biotech). SYTOX Green dead cell stain (Invitrogen) was added during the staining process to exclude dead or damaged cells. The labeled cells were sorted using a BD Influx flow cytometer. Fresh whole blood samples, pre-cooled to +4°C, were directly incubated with fluorescently labeled anti-human CD4 and CD8 antibodies (BD Biosciences). After a 15-minute incubation, samples were treated with BD FACS Lysing Solution to lyse red blood cells and reduce background noise. Finally, the processed samples were analyzed using a NovoCyte model 3000 flow cytometer (Acea Biosciences), and data collection and preliminary analysis were conducted using NovoExpress software.
Case presentation
A boy aged 10 years and 7 months was admitted to the hospital on March 6, 2022, with complaints of persistent cough for two months and fever for one day. The onset of his paroxysmal non-spasmodic cough, accompanied by purulent sputum of yellow-green color, began two months prior without any apparent triggers. Concurrently, he frequently exhibited symptoms of sneezing, nasal congestion, and purulent nasal discharge. Despite being diagnosed with bronchitis by an external hospital and receiving infusion treatments, his cough and purulent sputum did not improve, prompting further diagnosis and treatment at our facility. The child was delivered via cesarean section, weighed 4050 grams at birth, was breastfed, and received vaccinations on schedule. He had been frequently diagnosed with bronchitis since childhood, receiving multiple infusion treatments, and had a history of recurrent skin allergies, allergic rhinitis, warts, sinusitis, and otitis media. Additionally, he was treated at our hospital for a drowning incident in 2018. He has a healthy sibling with no notable family medical history.
Upon admission, physical examination revealed a heart rate of 90 beats per minute, respiratory rate of 32 breaths per minute, body temperature of 36.5°C, weight of 27 kg, and blood pressure of 90/60 mmHg. He was 137 cm tall, underweight, with no cyanosis around the mouth or face, and had clubbing. Auscultation of the lungs revealed widespread mid-wet rales with normal heart sounds. The complete blood count showed an elevated white blood cell count of 17.4 × 109/l, with 77.4% neutrophils, 18.8% lymphocytes, an extremely high C-reactive protein (CRP) level of 142.7 mg/l, and an erythrocyte sedimentation rate (ESR) of 64 mm/h. Immunological examination indicated a low percentage of CD3+CD4+ T cells, an increased percentage of CD3+CD8+ T cells and NK cells, elevated levels of immunoglobulins G, A, and M, and a significant increase in total IgE to 1837 IU/ml. With negative pathogen detection and autoantibody tests, liver and kidney functions, myocardial enzyme spectrum, electrolytes, and coagulation were all normal. An electrocardiogram, cardiac ultrasound, and abdominal ultrasound showed no abnormalities. Pulmonary CT indicated bronchiectasis with infection and multiple scattered nodular dense shadows in both lungs. Paranasal sinus CT showed uneven thickening of the mucosa in both maxillary sinuses, ethmoid sinuses, and sphenoid sinuses, especially notable thickening in the maxillary sinuses and significant thickening of the right nasal mucosa. Pulmonary function tests indicated mild restrictive ventilatory defect. Endoscopic examination of the bronchi revealed a large amount of yellow purulent secretion in the trachea and bronchial segments, causing blurred vision. Bronchoalveolar lavage fluid (BALF) analysis showed 80% neutrophils, 2% lymphocytes, and 18% epithelial cells. Pathogen metagenomics of BALF detected Haemophilus influenzae.
After admission, based on the pulmonary CT scan and pathogen metagenomics of BALF, the child received anti- infection treatment with cefoperazone-sulbactam. Notably, the child had been hospitalized in 2018 for drowning, and a pulmonary CT scan from September 3, 2018 (Fig. 1A) revealed extensive bronchiectasis with infection, but he did not undergo further follow-up after discharge. The current admission’s pulmonary CT showed bronchiectasis with infection and extensive small nodular shadows (Fig. 1B), considering the child’s history of recurrent respiratory infections, sinusitis, otitis media, and warts, suggesting a possible immune deficiency, leading to a recommendation for genetic testing.
Fig. 1
The chest CT of the patient showing bronchial dilatation with infection and diffuse nodular changes in both lungs
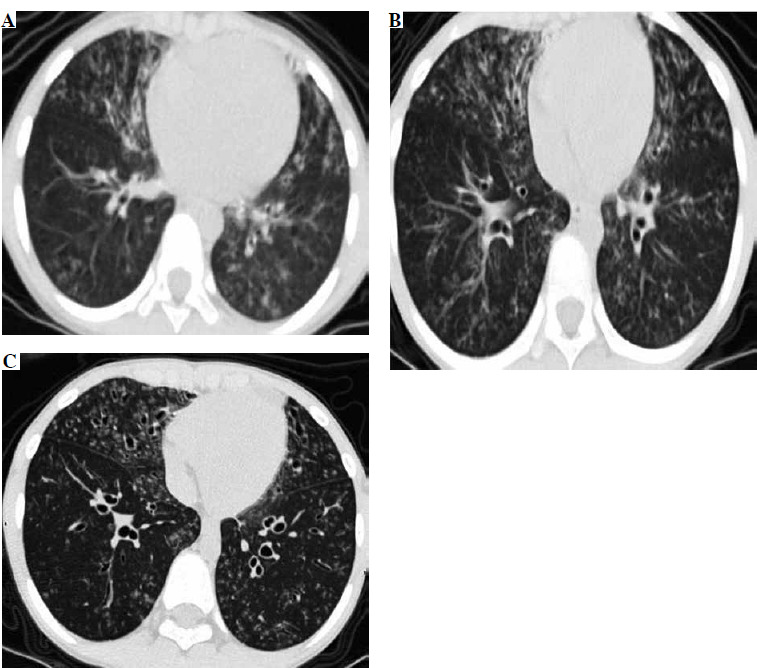
After 7 days of anti-infection treatment, follow-up bronchoscopy showed improved bronchial mucosal inflammation, no purulent secretion, significantly reduced cough and expectoration symptoms, and disappearance of lung signs. A high-resolution pulmonary CT scan on March 22, before discharge, showed significant improvement in bronchiectasis and lung infiltrates compared to previous findings (Fig. 1C).
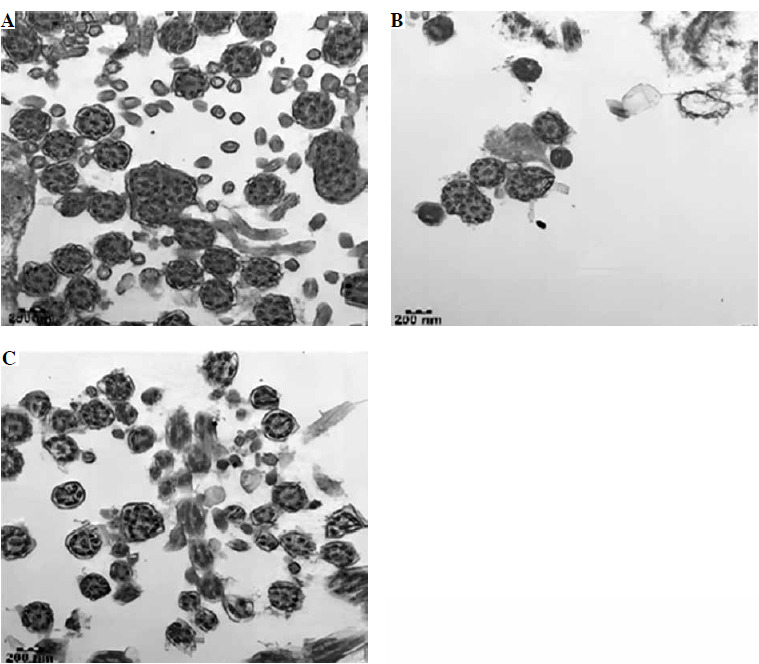
Following his discharge, the child’s condition remained stable. He was readmitted on April 13 for an endoscopic bronchial biopsy under electronic bronchoscopy, which was then analyzed using electron microscopy. Subsequent immunoglobulin tests indicated that the levels of immunoglobulins G, A, and M were still elevated, with a significant increase in total IgE. Lymphocyte subtyping on April 19 revealed abnormal distributions of T, B, and plasma cells. Electron microscopy of the mucosal biopsy on April 26 identified mild inflammatory changes and a few cilia with abnormal microtubule structures (Fig. 2).
Fig. 2
Results of the patient’s electron microscopy examination of bronchial mucosal biopsy during electronic fiber bronchoscopy
Over the following 18 months, the child made multiple visits to the Pediatric Hospital affiliated with Fudan University, where he was diagnosed with “primary immunodeficiency/IL2GR gene mutation, hyperimmunoglobulinemia, bronchiectasis with infection, sinusitis, otitis media, and skin warts”. To exclude primary ciliary dyskinesia, he underwent comprehensive medical examinations, including PET-CT, nasal ciliary tests, and further electron microscopy. Cultures from other hospitals repeatedly indicated colonization by Pseudomonas aeruginosa, for which nebulized tobramycin treatment was administered. During the COVID-19 and H1N1 influenza epidemics, the child was in good general condition, experiencing mild post-infection symptoms, primarily low-grade fever and cough, which resolved with symptomatic treatment.
Genetic analysis
With informed consent from the child and his family members, WES of peripheral blood samples from the child, his parents, sister, and uncle was conducted to identify genetic variants potentially associated with bronchiectasis. The sequencing, completed by the Beijing MyGenostics Medical Laboratory, did not reveal any pathogenic point mutations, large fragment deletions, or duplications highly associated with the clinical phenotype of bronchiectasis. However, a variant of uncertain significance in the IL2RG gene was identified at c.420A>T (p.R140S). The child’s mother and sister are heterozygous for this variant, while his father and uncle showed no variation at this site (Fig. 3).
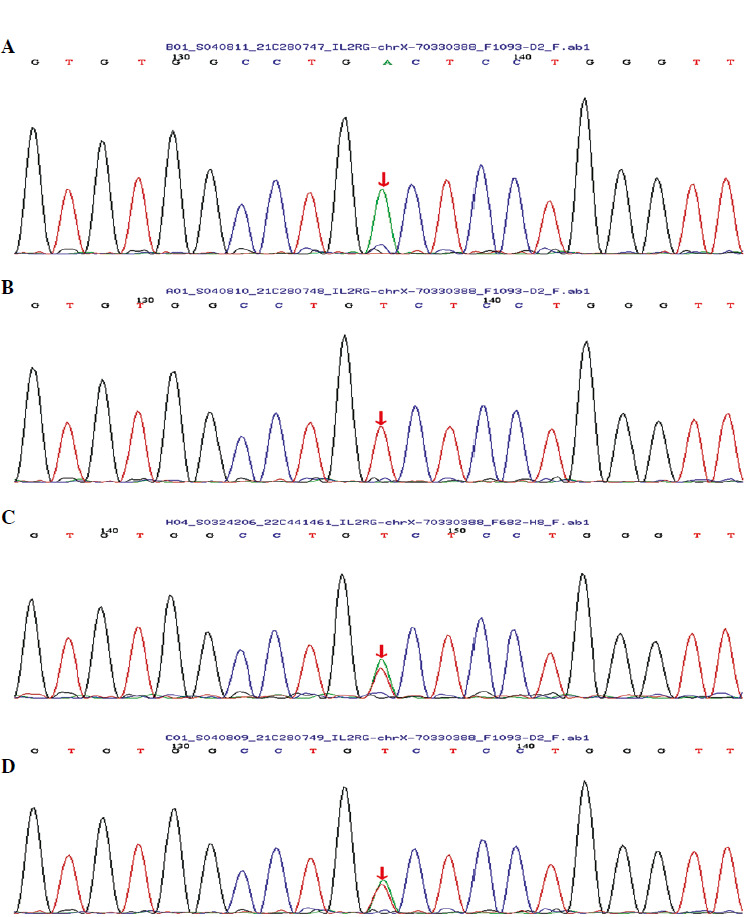
Discussion
The newly identified IL2RG gene variant in this study, c.420A>T (p.R140S), offers crucial insights for a comprehensive understanding of both primary combined immunodeficiency (CID) and X-linked combined immunodeficiency (X-CID), aiding in differential diagnosis. Although the patient exhibited manifestations such as delayed development (with thin stature, normal complexion without cyanosis, and clubbing of fingers and/or toes), severe lung infections (bronchiectasis with scattered nodular opacities in both lungs on CT scan), abnormal lymphocyte subtyping (abnormal distribution of T cells, B cells, and plasma cells), and the IL2RG gene mutation at the c.420A>T (p.R140S) site, the absence of markedly low T cell counts or clinical symptoms indicating T cell dysfunction led to a diagnosis of CID instead of X-CID. Notably, the pathogenic role of the IL-2Rγ chain protein encoded by IL2RG, located on Xq13.1, in regulating the differentiation, development, and maturation of T cells, NK cells, and B cells has been highlighted by Noguchi et al. [17] and Puck et al. [18] since 1993. Our report on this novel variant expands our understanding of the impact of IL2RG gene mutations in the spectrum of X-CID and CVID diseases, particularly in terms of atypical clinical presentations.
Mutations in the IL2RG gene lead to dysfunction of the encoded IL-2Rγ chain protein, affecting intracellular signaling in T cells, resulting in T cell dysfunction and severe T cell deficiencies. This defect impacts T cells and indirectly causes B cell dysfunction, ultimately leading to decreased cellular and humoral immunity, making the body prone to recurrent infections (Fig. 4). Amy P Hsu and others reported in 2015 a patient diagnosed with common variable immunodeficiency (CVID) who was later found to have a CD132 signaling defect due to an IL2RG mutation, revealing the potential atypical phenotypes caused by IL2RG mutations and the challenges they pose to clinical diagnosis [19, 20]. The case reported in this study also demonstrates similar complexity, where, despite the patient exhibiting typical CID symptoms (recurring bronchitis, otitis media, sinusitis, etc.), WES revealed an unreported novel IL2RG gene mutation. Currently, the ClinVar and HGMD databases list 446 IL2RG variants, predominantly missense, frameshift, nonsense, and splice site mutations, with missense mutations being the most common. The discovery of this case provides valuable new insights into the diversity of IL2RG variants and their relationship with different immunodeficiency disease phenotypes.
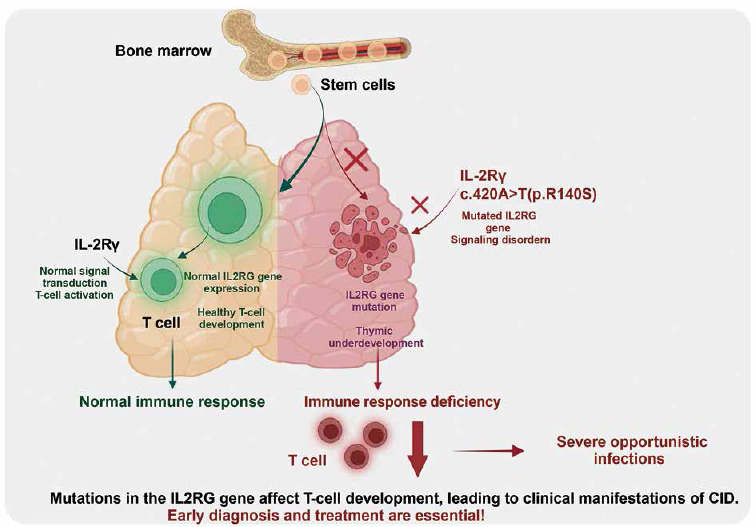
This case’s clinical and laboratory findings supported the diagnosis of CID, highlighting the complexity and diversity of immunodeficiency diseases. Notably, the total lymphocyte count in the child was average, but there was a decrease in CD4+ T cells, along with alterations in other specific immune cell subgroups, indicating a milder form of T cell dysfunction. Additionally, the high levels of total IgE, similar to cases reported by Brahim Belaid et al. [15], underscore the potential for functional abnormalities leading to humoral immunodeficiency despite average B cell counts.
The choice of treatment strategy is crucial for improving patient outcomes [21-23]. Early bone marrow transplantation has been shown to improve survival rates in patients with X-SCID significantly [24], while personalized treatment plans are necessary for CID or other atypical immunodeficiency diseases [4, 6]. This case underscores the importance of early diagnosis and timely intervention and the need to provide comprehensive treatment and management strategies for patients with rare diseases.
By identifying a novel IL2RG gene mutation (c.420A>T(p.R140S)), this study offers important insights into understanding atypical X-SCID and CID. This discovery directly affects clinicians in diagnosing and treating patients with specific immunodeficiency phenotypes. Firstly, it enhances understanding of the role of the IL2RG gene in immunodeficiency, emphasizing the importance of thorough genetic evaluation in the presence of atypical clinical symptoms. Secondly, the findings contribute to refining diagnostic criteria for X-SCID and CID, especially when patients exhibit non-typical clinical features. Lastly, for patients carrying specific IL2RG mutations, this study may promote more personalized treatment strategies, including early intervention and targeted treatment options, to optimize patient management and improve survival rates.
While this study provides valuable insights into the relationship between novel IL2RG gene mutations and atypical X-SCID and CID, it has certain limitations. Firstly, the study is based on a single case report, limiting our understanding of the mutation’s prevalence and distribution across different populations. Secondly, although a correlation between the new gene mutation and clinical phenotype was identified, there is a lack of detailed functional studies elucidating how this mutation affects the function of the IL-2Rγ chain protein and cellular signaling pathways. Additionally, the study did not encompass all potential genetic and environmental factors that could influence clinical manifestations, potentially limiting the interpretation of disease phenotypes.
Future research should focus on several key areas to overcome these limitations and further expand our understanding of atypical X-SCID and CID. Firstly, multicenter studies in a broader population are needed to assess the prevalence of the newly discovered IL2RG mutation and its distribution across different ethnicities and geographical regions. Secondly, detailed functional studies should be conducted to reveal how the mutation affects the function of the IL-2Rγ chain protein and how it interferes with immune cells’ normal development and function. Additionally, exploring other potential genetic and environmental factors associated with this mutation would help to understand the disease’s complexity and diversity comprehensively. Lastly, based on these findings, developing and testing new therapeutic approaches, especially for patients with atypical immunodeficiency phenotypes, could provide more effective treatment options and improve patient outcomes.