






Introduction
Portal hypertension (PH) is a clinical syndrome characterized by a pathological increase in pressure within the portal vein (PV) [1]. Based on the etiology of the disease and the location of the pathological substrate, PH can be divided into prehepatic, intrahepatic, and posthepatic type. Intrahepatic PH is caused by chronic liver disease. It can be further subclassified into three additional categories depending on the site of activity of the pathological substrate in relation to the sinusoids – presinusoidal, sinusoidal, and postsinusoidal PH. Presinusoidal PH is caused by liver diseases with the pathological substrate located proximal to the sinusoids (e.g., schistosomiasis, primary biliary cholangitis, and sarcoidosis). Sinusoidal PH, the most common form of PH, occurs in diseases that affect the liver parenchyma, cirrhosis being the main cause. In postsinusoidal PH, the pathological substrate is located distal to the sinusoids and is caused by obstruction of central veins (e.g., sinusoidal obstructive syndrome) [2]. Cirrhosis and schistosomiasis are responsible for more than 90% of cases of PH worldwide [3]. Additionally, PH can also occur due to obstruction of the portal flow before it enters the liver, for example, due to portal vein thrombosis (PVT). In such cases, PH is classified as prehepatic. When the obstruction happens after the blood exits the liver, for example in Budd-Chiari syndrome, PH is classified as posthepatic [2]. The gold standard for assessing the severity of PH is measurement of the hepatic venous pressure gradient (HVPG). This procedure is carried out through transjugular catheterization of the hepatic veins under fluoroscopic control, and it measures the difference between the wedged hepatic venous pressure (WHVP) and free hepatic venous pressure (FHVP), which serves as an indirect measure of portal pressure [1]. The normal value of HVPG ranges from 1 to 5 mmHg, values between 6 to 9 mmHg are considered subclinical PH, and values ≥ 10 mmHg are considered clinically significant portal hypertension (CSPH). Measurement of HVPG reliably reflects the severity of PH in patients with sinusoidal causes, while its values are normal or only slightly elevated in pre- and postsinusoidal forms of PH, where it lacks prognostic value but still holds diagnostic significance [1, 2, 4]. The classification and clinical features of different forms of PH are summarized in Table 1.
Table 1
Causes of portal hypertension. Adapted from [2]
Porto-sinusoidal vascular disease (PSVD) is a clinical entity that refers to patients with presinusoidal PH and was defined in 2019 on the proposal of the Vascular Liver Disease Interest Group (VALDIG) [5]. Prior to that, the term idiopathic non-cirrhotic portal hypertension (INCPH), which was introduced in 2011, was used, and before that, older names such as nodular regenerative hyperplasia [6], hepatoportal sclerosis [7], obliterative portal venopathy [8], and idiopathic portal hypertension [9] were used. The term INCPH encompassed patients with clinical signs of PH without cirrhosis and without PVT. However, it was found that such a definition had limitations because it did not include several categories of patients covered by the new definition. For example, patients with PH and histological signs typical of INCPH who had another (additional) chronic liver disease (e.g., non-alcoholic fatty liver disease – NAFLD, viral hepatitis, or alcoholic liver disease) were excluded from that diagnosis [10]. In addition, histological signs characteristic of INCPH could also be found in patients without signs of PH, so they were not included in the old definition of the disease, nor were patients with PVT, which was later determined to develop as a result of INCPH, so at the time of diagnosis, histological signs of INCPH and clinically confirmed PVT could coexist [11, 12]. Considering these conclusions and new insights, the VALDIG group proposed a new name and definition of this entity in 2019, encompassing the entire spectrum of disease, which will be further described in the following text.
Definition
Porto-sinusoidal vascular disease is defined as vascular liver disease characterized by the absence of cirrhosis on liver biopsy and the presence of histological lesions suggestive of this disease (such as obliterative portal venopathy, nodular regenerative hyperplasia, incomplete septal fibrosis), with or without the presence of PH, and possibly associated with the presence of other diseases that are linked to the development of histological changes characteristic of PSVD [5]. Specific and non-specific histological features used to define the presence of PSVD are presented in Table 2, while the diseases associated with the development of PSVD are presented in Table 3. The name of this clinical entity relates to the fact that the disease affects portal venules and/or sinusoids. As is evident, the criteria for this disease still include the absence of cirrhosis, along with histological signs characteristic of this disease. The presence of PH is no longer an inclusive criterion, and the presence of another chronic liver disease (e.g., NAFLD, viral hepatitis) is no longer an exclusive criterion for PSVD. The diagnosis of PSVD is easier to establish if one of the associated diseases that is linked to the pathogenesis of PSVD is present. Liver diseases associated with hepatic vein obstruction and specific microangiopathic diseases (shown in Table 4) are excluded from this definition [5].
Table 2
Definition of porto-sinusoidal vascular disease (PSVD). According to [5]
Table 3
Conditions associated with porto-sinusoidal vascular disease (PSVD). According to [5]
Table 4
Conditions excluded from the definition of porto-sinusoidal vascular disease (PSVD). According to [5]
Epidemiology and geographical distribution
The global incidence of PSVD is not well known, mainly due to its uneven geographical distribution, its relatively recent definition, and insufficient awareness within the hepatology community. Diagnosing patients who do not yet have PH is still challenging since they are asymptomatic in the early stages of the disease [13]. Although there are not many studies in Europe focusing on the incidence of PSVD, it is considered to be low. In one French study, the incidence of PSVD was 4% among 3600 liver biopsies [14]. A Spanish study showed an even lower incidence of 0.5% in its cohort of HIV-positive patients, and significant influence on the occurrence of this disease was attributed to didanosine treatment [15]. Men are more commonly affected, with a male-to-female ratio of 3 : 1 [16]. In North America, the prevalence is also low, around 3-7% in Canada and about 4% in the USA, with male predominance, and an average age range of 60-69 years [7, 9, 17]. The highest prevalence of PSVD was reported in India, accounting for 15-34% of all cases of PH [5], depending on the group of patients studied and diagnostic modalities that were used, whereas liver biopsy was not used in all the reported cases. Again, men were more frequently affected, with a male-to-female ratio ranging from 2 : 1 to 4 : 1. Although the incidence is declining, it remains high, implying that the disease is associated with lower socioeconomic conditions [5, 18-20]. In Japan, the prevalence and incidence of PSVD are declining, as 29-37% of patients who underwent surgery due to complications of PH in 1970s had idiopathic non-cirrhotic PH (PSVD), whereas only 11 new cases of PSVD patients are being surgically treated per year nowdays. This decline in incidence can be attributed to changes in health policies that have allocated more funding for research on rare diseases and the government’s coverage of treatment costs for these conditions [5]. PSVD in Japan occurs more frequently in women, with an average age range of 40-59 years. One possible reason is the higher prevalence of autoimmune diseases in women [21]. However, the female-to-male ratio has been decreasing, from 4 : 1 in 1984 to 2 : 1 in 2012 [22, 23].
Pathophysiology and etiology of the disease
The exact pathophysiology of this disease is still unknown; however, it is presumed to be caused by injuries and occlusion of the intrahepatic portal microvasculature, leading to increased resistance to portal blood flow and consequent presinusoidal type of PH [24]. Over 50% of patients have an associated disease (Table 3) that is linked to the development of histological changes in the liver typical of PSVD [5]. These diseases can be categorized into five groups: hematological disorders and thrombophilias, immunological diseases, infections, genetic diseases, and exposure to certain drugs.
Hematological disorders and thrombophilia
Studies have confirmed the role of microthrombi and platelet aggregation in the intrahepatic portal venules in the development of PSVD. This is supported by the fact that the most common histological finding in PSVD is thickening of the vessel wall and obliteration of the lumen in intrahepatic portal venules [5]. Consequently, deficiencies in protein C and factor V Leiden mutation have been associated with the development of this disease [25, 26].
Immunological diseases
It is assumed that 10% of patients with PSVD have some form of immune system disorder [5]. One study showed that 6% of patients with inflammatory bowel disease have PSVD, but it is unclear how much of this percentage is contributed by azathioprine, which is used in the treatment of these patients [27]. Other diseases that have been associated with PSVD include Bruton’s disease, hyper IgM syndrome, common variable immunodeficiency (CVID), and Felty’s syndrome [5]. The pathophysiological mechanism is also not clear, but it is presumed that intrasinusoidal T lymphocytes may cause portal vein or sinusoidal endotheliitis as the overexpression of lymphocyte activation genes was demonstrated in blood samples of patients with PSVD [28]. Furthermore, the elevation of macrophage activation markers (soluble CD163 and soluble mannose receptor) in patients with INCPH has been described, though it was less pronounced than in cirrhosis, suggesting that macrophage activation could play a role in the development of PSVD [29]. We could not find data to explain the pathophysiology of PSVD’s association with autoimmune hepatitis (AIH) as the paradigmatic autoimmune disease, but we assume that marked activation of the lymphoplasmacytic component in the liver might lead to the extension of inflammation to the terminal branches of the portal vein and finally result in terminal phlebitis. Again, development of PSVD in this clinical context might also be associated with the azathioprine therapy for AIH.
Infections
Research on rabbits has shown that translocation of intestinal bacteria into the PV could trigger an immune response that leads to histological changes similar to those seen in PSVD [30]. Chronic or recurrent infections could lead to mild portal inflammation, ultimately resulting in the histological picture of PSVD [31]. HIV infection has also been identified as an important etiological factor in the development of PSVD, partly due to antiviral therapy [31, 32].
Genetic diseases
Patients with the HLA-DR3 antigen have a higher likelihood of developing the disease [33]. Furthermore, there is a hereditary component associated with this condition. Mutations have been identified in the KCNN3 (potassium calcium-activated channel subfamily N member 3) gene, which encodes small conductance calcium-activated potassium channel 3, which is involved in the regulation of arterial and venous vascular tone by causing smooth muscle relaxation on activation. This mutation is transmitted in an autosomal dominant manner [34]. Another mutation has been found in the DGUOK gene, encoding deoxyguanosine kinase, which phosphorylates purine deoxyribonucleosides in the mitochondrial matrix and is required for replication of mitochondrial DNA. This mutation is transmitted in an autosomal recessive manner [35]. A recent study that sequenced the genome of four affected members of a Lebanese family with PSVD and two healthy members revealed an aberrant structure of the FCHSD1 (FCH and double SH3 domains 1) gene, leading to excessive expression of the FCHSD1 protein and enhanced activation of the mTOR signaling pathway [36]. Other genetic diseases associated with the development of PSVD include Adams-Oliver syndrome, Turner syndrome, familial obliterative portal venopathy, and cystic fibrosis [5].
Exposure to drugs
The medications most associated with PSVD are oxaliplatin, thioguanine, azathioprine, and didanosine [5]. A Dutch study from 2012 reported that 4% of patients with PSVD were HIV positive, and a potentially causative effect of therapy with didanosine and stavudine was suspected by the authors [32].
Diagnosis of porto-sinusoidal vascular disease
Histology
The diagnosis of PSVD according to the definition requires a liver biopsy, in which it is necessary to obtain an adequate tissue sample (> 20 mm in length, containing at least 10 portal tracts) [5]. Typical histological lesions in PSVD include obliterative portal venopathy, nodular regenerative hyperplasia, and incomplete septal fibrosis.
Obliterative portal venopathy or phlebosclerosis (for which the term “portal vein stenosis” has recently been proposed) includes a set of changes that involve narrowing of the PV branches up to complete obliteration of these branches and replacement with connective tissue. The vascular lesions are irregularly distributed across the liver parenchyma and vary greatly in the severity and extent [31, 37]. Nodular regenerative hyperplasia presents a perturbation in the lobular architecture leading to distortion of the normal relationship between portal and central areas. Hepatocytes undergone nodular regenerative hyperplasia represent poorly defined micronodular zones of the liver parenchyma in the absence of fibrosis and may reflect a more advanced stage of the same disease. Incomplete septal fibrosis can be recognized by thin portal-based fibrous septa incompletely demarcating the liver parenchyma into inconspicuous surround nodules without forming typical regenerative nodules as in cirrhosis. These fibrous septa are spread in periportal and centrilobular areas. Such a lesion may be difficult to separate from so-called incomplete septal cirrhosis that presents an advanced stage of chronic liver disease.
Due to all aforementioned histological features, liver biopsy is essential to exclude cirrhosis or other causes of portal hypertension and to identify PSVD features. It requires extensive knowledge and experience of the pathologist to confirm the clinical suspicion of PSVD [31]. The histological image of PSVD is shown in Figure 1.
Fig. 1
Histological features of the porto-sinusoidal vascular disorder in the liver parenchyma (authors’ own data). A, B) Obliterative portal venopathy/ phlebosclerosis. The key feature of porto-sinusoidal vascular disorder (PSVD), characterized by fibrous thickening and occlusion of the distal intrahepatic portal veins (PVs) within portal tracts, replaced with multiple thin-walled vascular channels. Also, distortion of the normal relationship between the portal tracts and central areas is common. The portal tracts are abnormally approximated to each other. Masson trichrome staining; magnification 200×, scale bar 200 μm. B) Immunohistochemical staining CD34 confirms complete absence of the PV and of proliferation of neovessels in portal tracts with an increase in the total number of thin-walled vascular channels, another major feature of PSVD. Magnification 200×, scale bar 200 μm. C) Incomplete septal fibrosis. The vascular changes are accompanied by portal and periportal fibrosis of varying extent, with slender portal-based fibrous septa demarcating the liver parenchyma into inconspicuous nodules. Masson trichrome staining; magnification 100×, scale bar 200 μm. D) The hepatocytes may undergo complete or partial nodular regenerative hyperplasia. Poorly defined zones with widened hepatic plates alternating with localized parenchymal compression accentuated with Gomori staining. Magnification 400×, scale bar 100 μm. HA – hepatic artery

Hepatic venous pressure gradient
Hepatic venous pressure gradient is the gold standard for assessing the severity of PH [1]. In patients with PSVD who have CSPH, the HVPG value mostly does not exceed 10 mmHg. The reason is the presinusoidal component in the development of PH, which does not reflect on the WHVP, and frequent presence of intrahepatic venous shunts [31].
Ultrasound and elastography
Ultrasound can mainly display the consequences of PH (e.g., ascites, portosystemic collaterals, splenomegaly), as well as one of the main complications of PSVD, which is PVT. Regarding liver stiffness measurements (LSM) obtained by transient elastography (TE), they are generally below the range of cirrhosis, despite the nodular liver surface on imaging as seen in some patients, and median LSM of 8 kPa was reported by TE. Although some patients may show values above 10 kPa, they are still far below the threshold of 25 kPa used to noninvasively predict the presence of CSPH [38, 39]. On the other hand, spleen stiffness (SS) is significantly elevated, reflecting the presence of PH, and SS measurement should be considered in the case of clinical suspicion of PSVD [31, 40].
Contrast-enhanced ultrasound
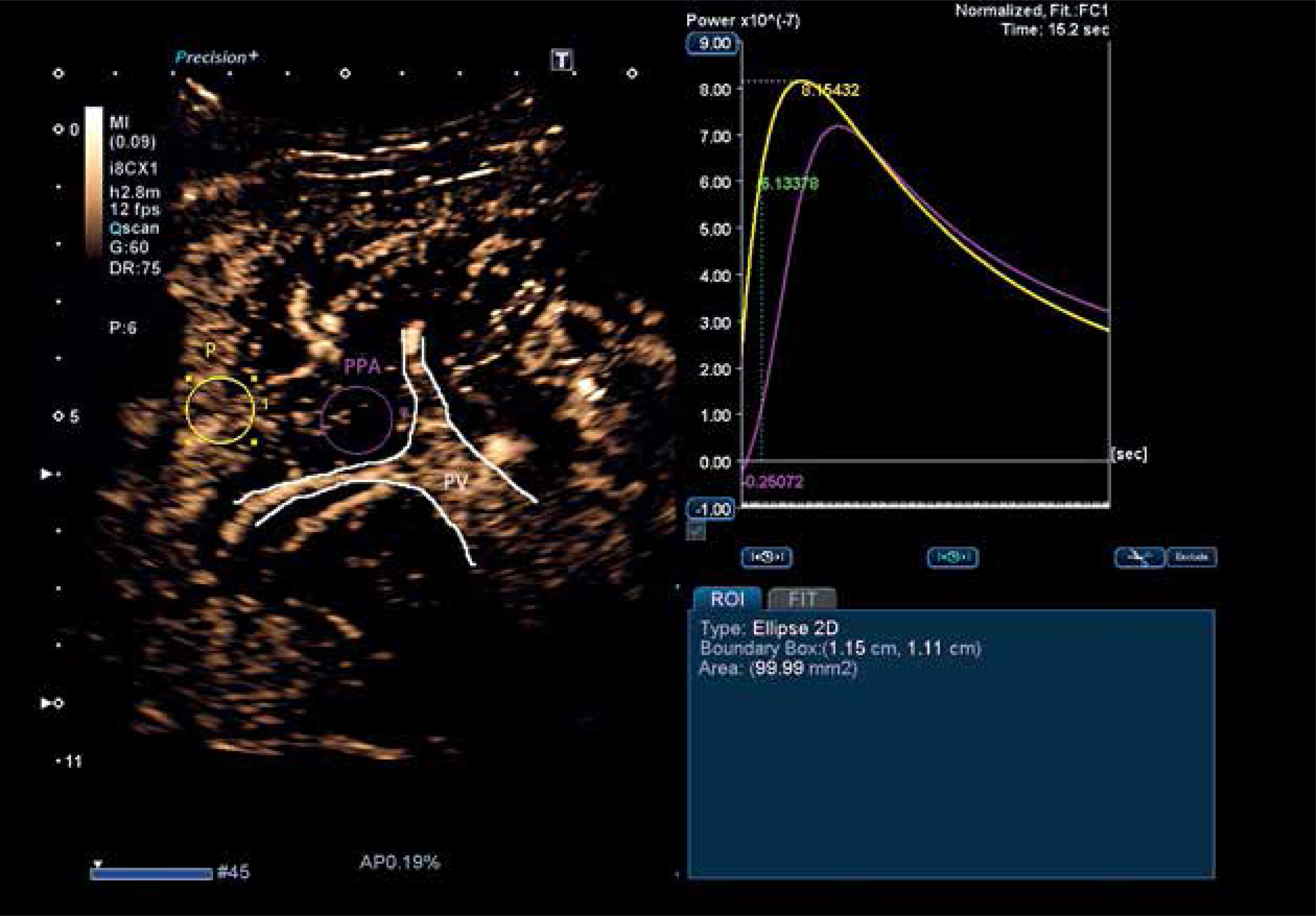
There is not a large amount of data on the use of contrast-enhanced ultrasound (CEUS) for diagnosing PSVD. However, a Japanese study used CEUS and time-intensity curve (TIC) analysis and noted that contrast enhancement occurred with a delay in periportal areas in patients with PSVD [41]. An example of a CEUS image and TIC analysis is shown in Figure 2.
Fig. 2
Contrast enhanced ultrasound with time intensity curve analysis in a patient with porto-sinusoidal vascular disorder (PSVD) (authors’ own data). The yellow circle is placed on the liver parenchyma, and the yellow curve represents the analysis of parenchymal intensity over time. The purple circle is placed in the periportal area, and the purple curve represents the analysis of periportal area intensity over time. The purple curve shows that there is delayed contrast enhancement in the periportal area compared to the rest of the liver parenchyma, which is consistent with the findings of Maruyama H et al. (Hepatol Int 2012 [41]). P – parenchyma, PPA – periportal area, PV – portal vein
Multislice computed tomography and magnetic resonance
The diagnosis of PSVD can be suspected based on several radiological features. Primarily, these include the presence of focal nodular hyperplasia (FNH)-like lesions in the liver parenchyma and “hyperenhancement” in the hepatobiliary phase using gadolinium as a contrast agent in magnetic resonance (MR) imaging. There is also a simple radiological scoring system that can differentiate PSVD from liver cirrhosis effectively. It observes five radiological features, namely: pathology of the intrahepatic portal system, pathological liver morphology (defined as peripheral parenchymal atrophy and compensatory hypertrophy of central segments and segment I), atrophy of segment IV, FNH-like lesions, and “hyperenhancement” of the periportal area. Each of these features carries 1 point, while 1 point is deducted for the presence of nodular liver parenchyma. According to this scoring system, patients with PSVD had a median score of 2 points, while patients with cirrhosis had –1 point. The optimal cut-off was 1 point according to Youden’s index, with an AUROC of 0.913 [42].
Clinical presentation and prognosis
According to the clinical presentation, patients can be divided into two groups – those with present clinical signs of PH and those without. Patients who have not developed signs of PH may have mildly elevated liver enzymes (elevated AST or ALP up to 2 times above the upper limit), leading to liver biopsy, which can confirm PSVD. It has been found that PSVD is present in 19% of cases of cryptogenic liver disease [11]. Patients with PSVD usually do not have symptoms until they develop complications of PH. The disease has a slow progression [5]. The most common laboratory finding after the appearance of PH signs is thrombocytopenia. Transaminases, ALP and GGT may be elevated, but only moderately (up to 2 times above the upper limit) in up to 80% of patients [5]. Liver function usually remains preserved, with most patients having normal levels of albumin and bilirubin [31]. 20-40% of patients with PSVD initially present with variceal bleeding, while ascites (present in 20-50% of cases) and encephalopathy are rare initial symptoms. It has also been found that esophageal varices develop in 20% of patients on average 10 years after diagnosis [45]. PVT develops in 30-40% of patients with PSVD and PH, with a median of 5 years after diagnosis, but complete obstruction occurs in only a third of patients [5, 45, 46]. The development of hepatocellular carcinoma (HCC) in this disease is rare, so screening for HCC is not currently recommended in these patients [5, 13, 31, 47]. A Dutch study reported a 78% five-year survival rate without transplantation in their cohort, with only 13% of patients dying from liver-related causes, while 6 (9.7%) patients underwent liver transplantation [47]. Another study showed a three-year survival rate of 84.4%, with liver-related mortality at 3.9% [48]. A recent study, published this year, followed 33 patients with PSVD over a period of 16 years. The survival rates after 1, 5, and 10 years were 94%, 87%, and 58%, respectively. However, out of the 14 deaths during the mentioned period, only 4 (12.1%) were directly related to decompensated liver disease. The risk of PVT after 1, 5, and 10 years was 16%, 25%, and 48%, respectively [49]. The presence of focal nodular hyperplasia-like lesions was reported in 30% of patients with PSVD [42]. On the other hand, no study has analyzed the prevalence of PSVD in patients with FNH, as performing the liver biopsy and/or HVPG measurements in all patients with incidentally discovered FNH would probably not be justified. However, given the data about the prevalence of FNH in PSVD, we believe that assessment of both liver and spleen stiffness might be justified in patients with FNH, and if the results are suggestive of PH without cirrhosis, liver biopsy should be considered.
When to suspect porto-sinusoidal vascular disease
There are several clinical conditions where it is advisable to consider PSVD as a differential diagnosis. Primarily, these include patients with esophageal varices, variceal bleeding, or developed portosystemic collaterals in the absence of chronic liver disease or when there is a discrepancy between evident signs of PH and only mild liver damage [5]. Furthermore, PSVD may be suspected in patients with thrombocytopenia (< 150 × 109/l) and splenomegaly (> 13 cm). Some of these patients are also undergoing hematological evaluation; in fact, 8-17% of patients with PSVD have an associated myeloproliferative neoplasm [16, 32, 45]. Chronic and unexplained elevation of liver enzymes (ALT, ALP up to 2 times above the upper limit) can also be a reason to suspect PSVD, as well as the presence of diseases and conditions that are related to the development of histological changes characteristic of PSVD based on the etiopathogenesis (Table 3). Finally, in cases of PVT, PSVD should also be considered, as PVT occurs in 30-40% of patients with PSVD. However, in some situations, caution is required since the histological differentiation between intrahepatic PVT as part of PSVD and idiopathic PVT is not yet fully defined [5, 45, 46].
Treatment
Treatment is based on preventing complications of PH. Currently, there is no reliable method for screening these patients. Patients with esophageal varices are treated according to guidelines for cirrhosis, which includes primary prophylaxis with non-selective β-blockers (NSBB) and variceal ligation in case of intolerance or contraindication to NSBB [5, 38]. Transjugular intrahepatic portosystemic shunt has shown equally good results as in cirrhotic patients [50]. Indications for liver transplantation are the same as in cirrhosis [5]. Some experts propose anticoagulant prophylaxis in these patients, as it has been shown that portal venules were obliterated in 100% and large PV branches in 67% of explanted livers of patients with PSVD who underwent liver transplantation [51]. Furthermore, the development of PSVD is associated with thrombophilias, and most importantly, PVT is a common complication occurring in 13-45% of patients with PSVD [45, 47, 51]. However, since there are still no randomized clinical trials on this topic and the risk of bleeding is still unclear, anticoagulant prophylaxis is not indicated according to guidelines, except in cases of existing PVT or thrombophilia with a high risk of PVT [5]. It is also noteworthy that anticoagulant therapy leads to PV recanalization in only half of the patients [45].
Unresolved issues and personal view
Although the definition of PSVD represents much improvement in comparison to the previous nomenclature, it may not completely cover all possible clinical situations. For example, incomplete septal fibrosis might be a result of a concomitant chronic liver disease, in which case it would be difficult to estimate the causality of PH if already present, i.e., to consider PSVD as the cause of PH if other indicative histological changes are lacking. On the other hand, the presence of incomplete septal fibrosis alone, without evidence of any other chronic liver disease after thorough clinical work-up, might be in our opinion considered as a sign of PSVD, especially if accompanied with PH. Liver biopsy is mandatory to diagnose PSVD, and it would be wrong to rely on a non-invasive result that excludes cirrhosis, if it is coupled with the presence of one specific sign of PH (such as esophageal varices). Portal hypertension in the absence of cirrhosis may be caused by other pre- and post-sinusoidal conditions, as previously pointed out, and liver biopsy is needed to rule them out. There remains a lot of work to draw a clear picture of the pathophysiological cascade responsible for the development of PSVD in the context of the associated clinical conditions, which might reveal potential therapeutic targets, as well as to define the potential usefulness of anticoagulant prophylaxis, as already pointed out.
Conclusions
Recently, clear histological and clinical criteria for diagnosing PSVD have emerged. However, this disease is still poorly recognized because symptoms usually appear only after complications of PH occur [5]. The exact pathophysiology of this condition remains unknown, but it is assumed that injuries and occlusion of intrahepatic portal microvasculature are the cause, leading to increased resistance to portal blood flow and consequent presinusoidal PH [24]. Half of the patients also have an associated condition that can contribute to the development of PSVD [5]. In addition to clear histological criteria, other non-invasive diagnostic methods for PSVD are being increasingly developed to diagnose patients early, before the disease progresses to severe stages. 20-40% of patients with PSVD initially present with variceal bleeding [45]. Another major complication of this disease is PVT, which can affect up to 45% of patients [45, 47]. So far, no additional risk of developing HCC has been observed in these patients [5, 13, 31, 47]. Treatment is based on managing the complications of PH, while anticoagulant prophylaxis is currently not indicated in the absence of randomized clinical trials on this subject [5]. Although this disease is becoming more recognized, further research is needed to enable early diagnosis, develop screening methods and intervals, and find ways to slow down its progression.