






Spontaneous coronary artery dissection (SCAD) is a non-traumatic and non-iatrogenic separation of the coronary arterial wall with intramural hematoma formation, leading to acute coronary syndrome, especially among young and middle-aged women [1]. Pre-disposing factors are fibromuscular dysplasia (FMD), pregnancy and postpartum period, multiparity, connective tissue disorders, and hormonal therapy [1]. In some cases other vascular beds may also be affected by spontaneous dissections [2]. Rare causal pathogenic genetic variants occur in a minority of SCAD patients, mostly in association with known concomitant connective tissue diseases, such as Ehlers-Danlos, Marfan, Loeys-Diez syndrome or autosomal dominant polycystic kidney disease (ADPKD) [1].
We present the case of a 39-year-old female with a history of hypertension and autosomal dominant polycystic kidney and hepatic disease (Figure 1 A), who was admitted to the hospital for recurrent chest pain lasting for last 6 h that occurred 2 weeks after cesarean section. The pregnancy was terminated by cesarean section at the 36th week due to preeclampsia. On admission, blood pressure was normal (135/90 mm Hg) and the ECG showed ST depression in leads V1-V3 with concomitant T-wave inversion (Figure 1 B). No right ventricle dilatation was found on echocardiography. However, as the patient had recently been pregnant, and the D-dimer level was mildly elevated (1.54 μg/ml) pulmonary embolism was first considered as the cause of the chest pain. Computed tomography (CT) angiography revealed normal pulmonary arteries. Then, based on increased troponin levels, non-ST-elevation myocardial infarction was diagnosed and the patient was referred for coronary angiography 12 h after chest pain onset.
Figure 1
A – Multiple renal and hepatic cysts on computed tomography. B – Electrocardiography on admission. C – Spontaneous coronary artery dissection in mid-distal segment of left circumflex artery (arrows) in coronary angiography. D – Coronary CT angiography confirming SCAD healing. E – Cervical CT angiography with carotid fibromuscular dysplasia. F – Cardiac MRI showing transmural late gadolinium enhancement in left circumflex territory (mid inferolateral segment of the left ventricle)
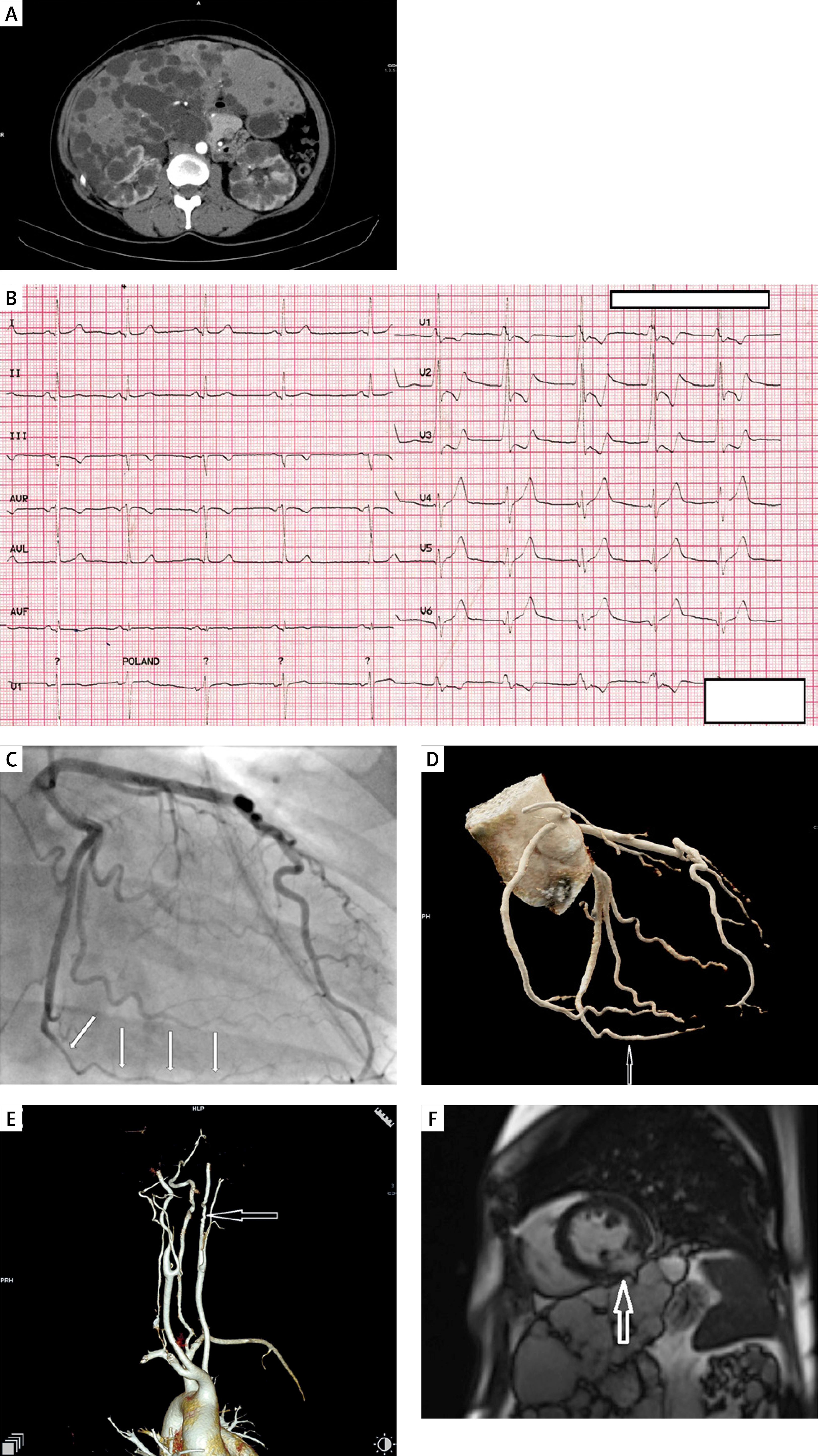
Coronary angiography showed long stenosis of the left circumflex artery with preserved TIMI 3 flow. After nitroglycerine intracoronary administration and vasospasm exclusion, based on typical angiographic appearance, type 2b spontaneous coronary artery dissection of the left circumflex artery was diagnosed (Figure 1 C). No intravascular imaging was performed. SCAD was managed conservatively. At discharge, echocardiography revealed mild hypokinesia of the basal and middle segment of the inferior wall, with left ventricle ejection fraction (LVEF) of 62%. One-year follow-up was uneventful. Control brain-to-pelvis CT angiography was performed then and revealed complete healing of the SCAD and bilateral carotid fibromuscular dysplasia (Figures 1 D, E). Cardiac MRI presented transmural late-gadolinium enhancement in mid inferolateral segment of the left ventricle, confirming myocardial infarction with LVEF of 55% (Figure 1 F).
To our knowledge, this is the first report of pregnancy-associated SCAD in a patient with autosomal dominant polycystic kidney disease. All few previously reported cases were not associated with pregnancy [3–5]. In patients with acute chest pain and concomitant ADPKD, SCAD should always be considered as the cause of acute coronary syndrome. In the present case, pulmonary embolism was initially considered; it delayed coronary angiography and accurate diagnosis. It also should be emphasized that triple “rule-out” CT angiography (for coronary, pulmonary arteries and aorta assessment) may have also limited specificity in SCAD cases, as dissections often affect distal coronary segments.
According to the guidelines, conservative management is recommended in low-risk patients with preserved coronary flow followed by screening for extracoronary vascular abnormalities such as FMD and aneurysms. In the case of comorbidities such as diabetes and severe hyperlipidemia, atherosclerotic etiology must be considered in differential diagnosis of patients with ADPKD. In equivocal cases, intracoronary imaging may support the diagnosis and facilitate the decision on revascularization.