






Primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma (PCAECTCL) was first described by Berti et al. as a distinct disease entity with distinctive clinical, histopathological, and immunophenotypic features [1]. PCAECTCL is currently classified as a rare subtype and provisional entity according to the latest World Health Organization-European Organization for Research and Treatment of Cancer (WHO-EORTC) classification [2]. Expression of CD8+/CD4- phenotype is a typically immunohistochemical finding in all cases, but some rare cases with aberrant immunophenotypes may be diagnostic and therapeutic challenges.
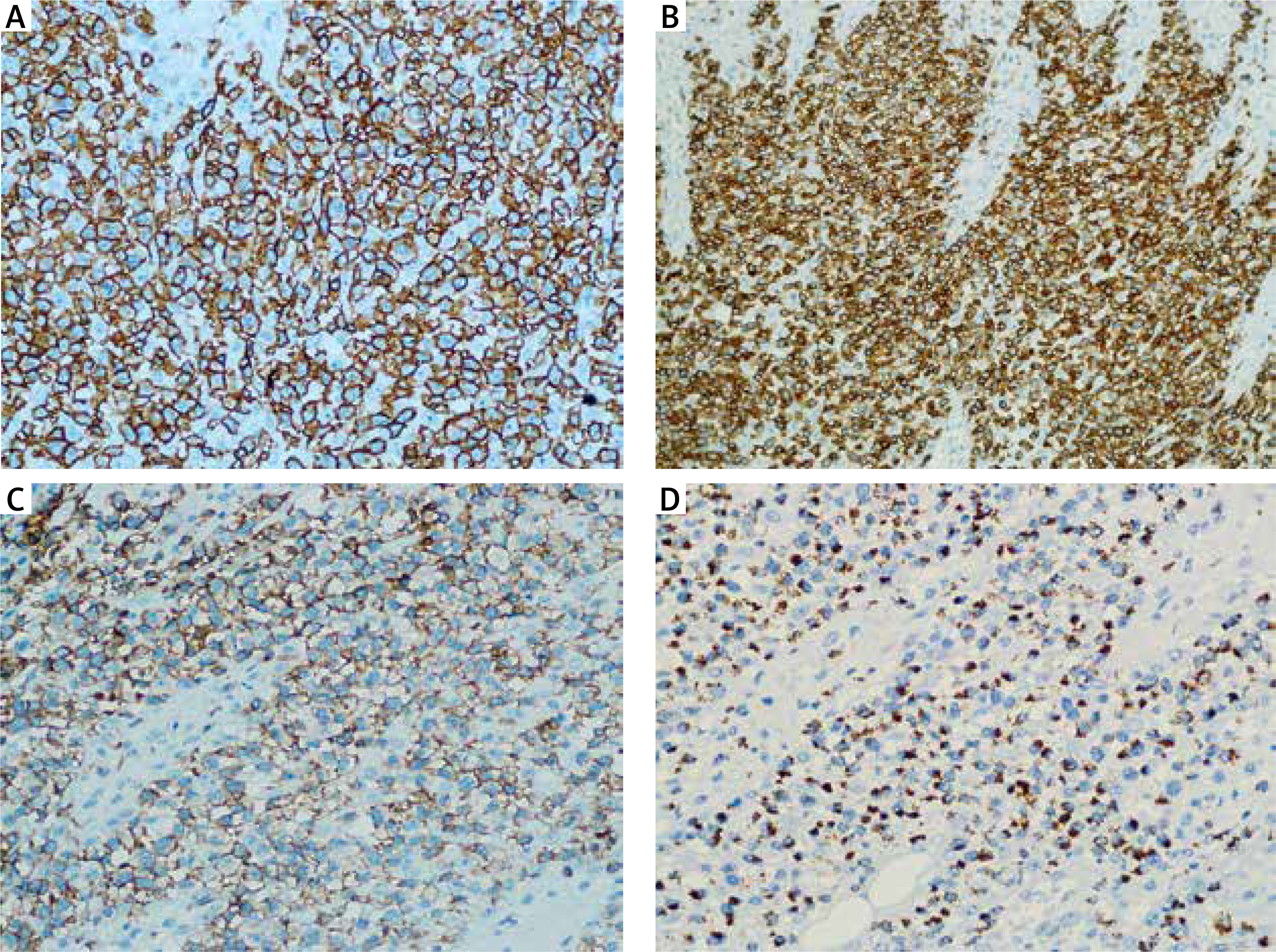
A 55-year-old Chinese man presented with generalized erythema, multiple reddish papules, and plaques on the trunk and proximal extremities for 3 months. He did not complain of fever, fatigue, weight loss, or lymphadenopathy since the onset of the illness. Physical examination revealed multiple infiltrated plaques and tumours with scale distributed over the trunk and extremities, some of which were ulcerated (Figure 1). A skin biopsy revealed a dense proliferation of medium- to large-sized atypical cells in the superficial dermis with irregular nuclei and prominent epidermotropic infiltration (Figure 2). Necrotic keratinocytes were not observed. Immunohistochemistry studies showed that atypical lymphocytes were positive for CD3, CD5, CD7, CD8, CD56, TIA-1, Bcl-2, and Ki-67(60%) (Figure 3), and negative for CD4, CD20, CD79a, CD10, CD30, and MUM-1. Epstein-Barr encoding region (EBER) in situ hybridization was negative. T-cell receptor-γ rearrangement demonstrated T-cell origin and monoclonality. Studies for evaluation of the extension of disease with hemogram, blood tests including lactate dehydrogenase (LDH), human T-lymphotropic virus (HTLV) type I and II, human immunodeficiency virus (HIV), peripheral blood smear, bone marrow biopsy, as well as computed tomography (CT) scan did not find any alterations. Based on a combination of clinical, histological, immunohistochemical, and genetic profiles, a diagnosis of PCAECTCL was made.
Figure 1
A–D – Clinical presentation of the patient. Multiple infiltrated plaques and tumours with scale distributed over the trunk and extremities, some of which were ulcerated
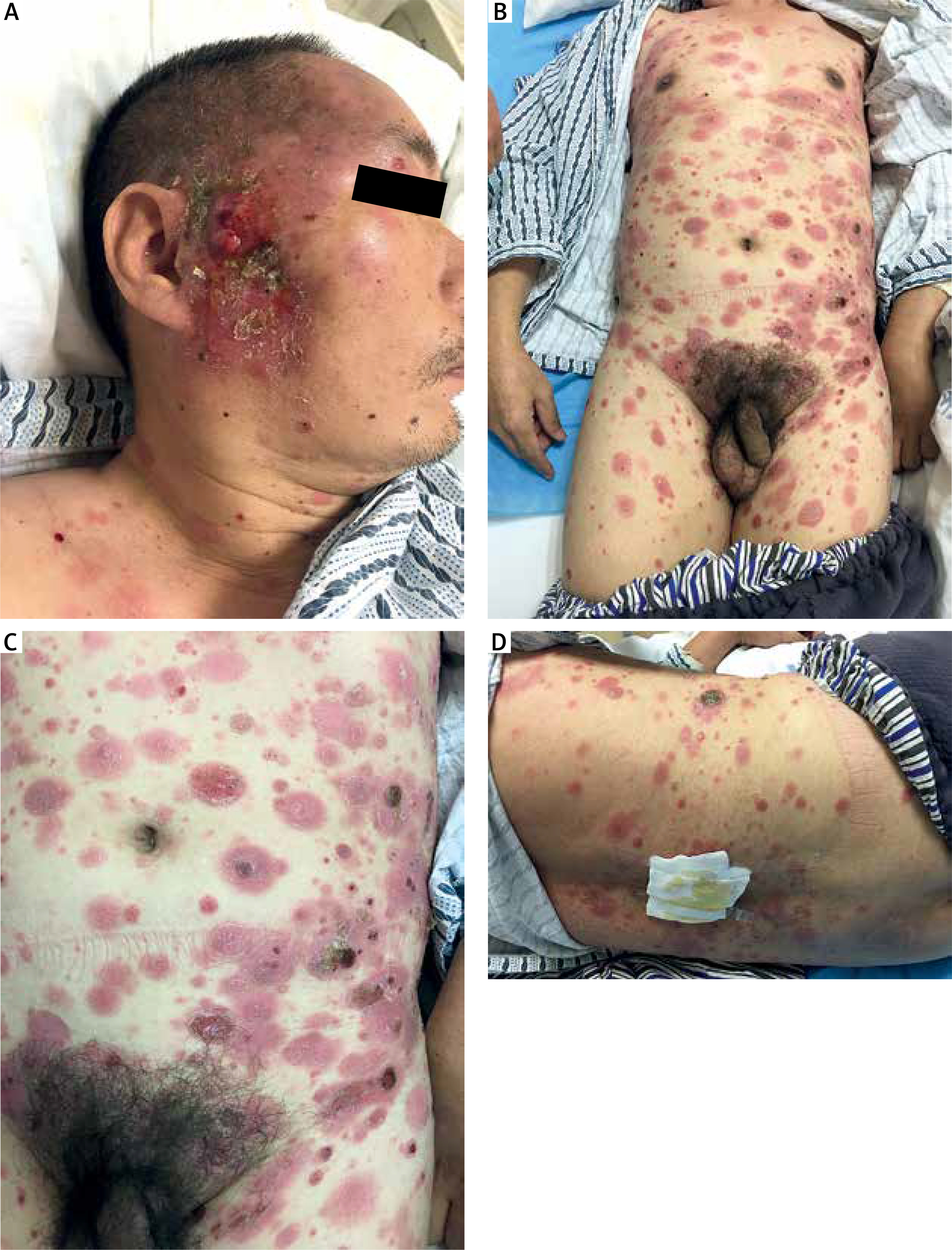
Figure 2
Histopathological result revealed a dense proliferation of medium- to large-sized atypical cells in the superficial dermis with irregular nuclei and prominent epidermotropic infiltration. Necrotic keratinocytes were not observed (A – H&E, ×100; B – H&E, ×400)
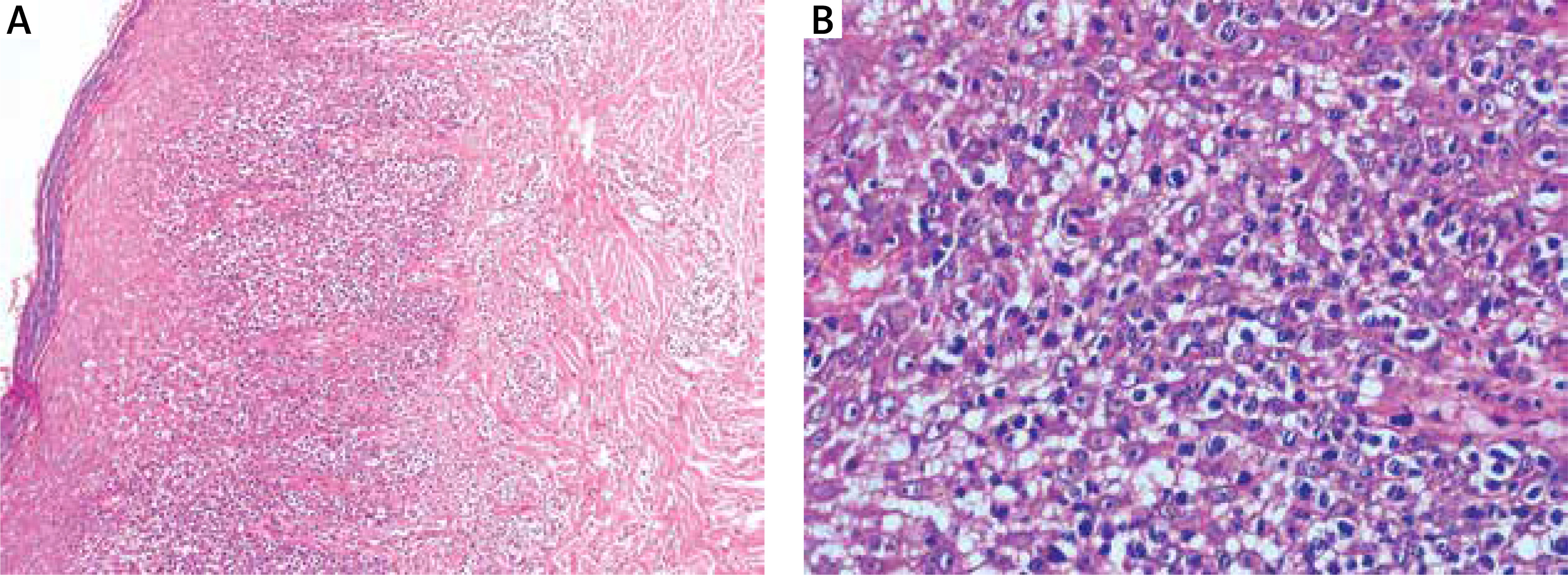
Figure 3
Immunohistochemistry showed positive staining for CD3 (A – Sp, ×400), CD8 (B – Sp, ×400), CD56 (C – Sp, ×400), and TIA-1 (D – Sp, ×400)
The patient was initially treated with gemcitabine, resulting in partial response, but he relapsed 5 weeks later. Despite referral for polychemotherapy of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) in the haematology department, he died of multiple metastases 15 months after the diagnosis.
PCAECTCL usually manifests as rapidly progressive, widespread papules, plaques, and tumours, often ulcerated [1, 3, 4]. Pyoderma gangrenosum-like lesions or superficial hyperkeratotic patches and plaques can be seen in some variable cases [5, 6]. PCAECTCL is aggressive in its clinical behaviour with rapidly progressive skin lesions and systemic symptoms. Despite the absence of precursor symptoms, the neoplastic lymphocytes often spread to extracutaneous sites. Our patient was living for only 15 months after the diagnosis.
Histologically, prominent epidermotropism resembling a pagetoid or linear pattern is seen throughout the different stages of the disease [1, 2, 7]. Pleomorphic lymphocytes present with nodular or diffuse infiltration in the superficial dermis. Cutaneous adnexal and blood vessels are commonly involved [1, 2, 7, 8]. The immunohistochemical study is important in diagnosis but expresses variably [1, 2, 4, 7]. Neoplastic T cell constantly exhibits CD8+/CD4- phenotype. Cytotoxic granules, such as TIA1, granzyme B, and perforin are always positive [1, 2]. Ki-67 proliferation index is often over 50%. CD2, CD5, CD7, and CD15 showed variable expression profiles, and pan-T-cell markers may be lost [9]. CD56 is a typical surface marker of natural killer (NK) cells. In the original description by Berti et al., CD56 was consistently negative [1], but CD56+ cases classified as PCAECTCL have been described in the literature [7]. The course of these patients was rapidly progressing followed by death, despite the use of aggressive multiagent chemotherapy regimens. Therefore, systemic spread may be attributed to the CD56+ phenotype, which may be associated with homing to extranodal sites [7].
CD56 expression was also observed in our case. Considering the aggressive course of the disease and resistance to treatment, the results of our study confirmed the speculation of Santucci et al. concerning the distinctive CD56 expression of neoplastic cells [7]. However, these observations were extremely rare and not consistent in all reported cases. Thus, we cannot use only one immunohistochemical maker as the criteria for diagnosing or predicting disease progression. Kato et al. reported a case showing genomic instability, which is consistent with the progression from localized cutaneous lesions toward more advanced systemic metastasis [10]. Accordingly, we speculated that genomic instability may also be responsible for the rapidly progressive features and CD56 expression that appeared in our case.
Considering the feature of epidermotropic CD8+ T lymphocytes, PCAECTCL needs to be differentiated from CD8+ cytotoxic MF, Lymphomatoid papulosis (Lyp) type D, and cutaneous γ/δ T-cell lymphoma (CGDTCL). Patients with MF present with a staged clinical presentation and behaviour, which are different from patients of PCAECTCL with generalized plaques and tumours at the onset of their disease [11]. Lyp type D is characterized by the typical “waxing and waning” of papules and small nodules [12]. Distinction from CGDTCL is achieved mainly by demonstration of expression of γ/δ and negativity for α/β [13].
Treatment for this disease remains a challenge because of poor responses to various therapeutic strategies and a high incidence of relapses [4, 13]. Multiagent chemotherapy such as CHOP regimen or hyper-cyclophosphamide, vincristine, adriamycin, and dexamethasone (hyper-CVAD) regimen is only responsive in a few cases [3]. Allogeneic hematopoietic stem cell transplantation (HSCT) might be considered in unresponsive cases or followed after multiagent chemotherapy [1, 14]. Recent literature showed the significance of aberrant activation of the JAK-STAT pathway in PCAECTCL, suggesting the possibility of JAK inhibitors such as ruxolitinib, but its efficacy in CTCL has not been established yet [15].
In conclusion, this is the first published case of PCAECTCL with aberrant CD56 expression in the Chinese population. Immunophenotypes may express variably in some rare cases of PCAECTCL. We reported this rare case to increase the awareness of this rapidly progressive disease and remind the possibility of variable immunophenotype in the rare subtype.