









Ferroptosis
In 2012, ferroptosis was first proposed by Dixon as a new type of cell death characterized by its dependence on iron. It belongs to a non-apoptotic mode of cell death and is characterized by the accumulation of reactive oxygen species (ROS) and the generation of oxidative damage in lipids [1]. Ferroptosis does not exhibit the typical features of traditional apoptosis (Table I), such as cell shrinkage, chromatin condensation, the formation of apoptotic bodies, and cytoskeletal breakdown. In contrast to autophagy, ferroptosis does not involve the formation of classical closed double-membraned structures (autophagic vacuoles) [2].
Table I
Compact characteristic of ferroptotic cell death (based on [2])
[i] p53 – tumour protein p53, GPX4 – glutathione peroxidase 4, TFRC – transferrin receptor 1, SLC7A11 – solute carrier family 7 member 11, NRF2 – nuclear factor erythroid 2-related factor 2, NCOA4 – nuclear receptor coactivator 4, HSPB1 – heat shock protein family B (small) member 1, ACSL4 – acyl-CoA synthetase long-chain family 4, FSP1 – ferroptosis suppressor protein 1.
Morphologically, ferroptosis is primarily characterized by pronounced mitochondrial shrinkage with increased membrane density, cristae circularization, and reduction or disappearance of mitochondrial cristae, which distinguishes it from other types of cell death [3]. Among the biochemical features of ferroptosis, intracellular glutathione (GSH) depletion, reduced activity of glutathione peroxidase 4 (GPX4), and generation of a large quantity of reactive oxygen species (ROS), which promote ferroptosis, could be mentioned [1–3].
Genetically, ferroptosis is a process regulated by many genes, including p53, GPX4, TFRC (transferrin receptor), SLC7A11 (solute carrier family 7 Member 11), and ACSL4 (acyl-CoA synthetase long chain family member 4). Ferroptosis mainly involves genetic alterations in iron homeostasis and lipid peroxidation metabolism [2].
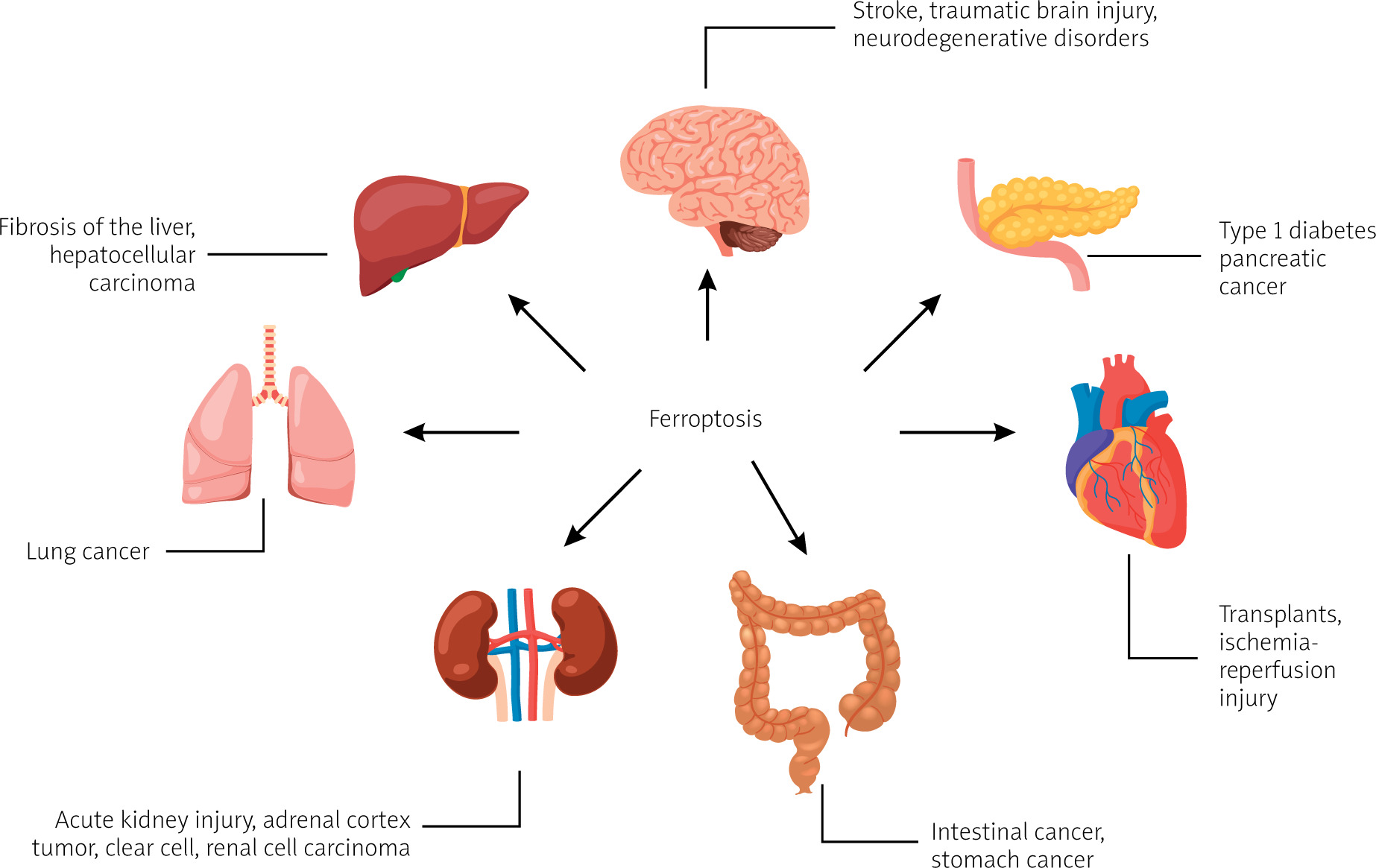
The literature research showed that ferroptosis plays a significant regulatory role in the occurrence and development of many diseases, and it has become a subject of research for the treatment and improvement of the prognosis of related diseases (Figure 1 [4, 5]), including ischaemic organ damage, neurodegenerative diseases, and cancer [2].
Figure 1
Diseases and disorders in which ferroptosis is observed, based on [4, 5]. Created with BioRender.com
Ferroptosis induction and prevention
RSL3 (RAS-selective lethal small molecule 3), a ferroptosis inducer, directly interacts with GPX4 and inhibits its activity, thereby reducing the antioxidant capacity of cells and accumulating reactive oxygen species (ROS), ultimately leading to ferroptosis [6]. Scientific literature suggests that the Xc-GSH-GPX4 pathway is the primary system preventing ferroptosis. If any stage of the process is disrupted, GPX4 activity decreases, leading to the accumulation of intracellular peroxides and contributing to the intensification of ferroptosis [6].
Ferroptosis suppressor protein 1 (FSP1), formerly known as apoptosis-inducing factor 2 (AIFM2), is a phosphor-nicotinamide adenine dinucleotide (NADP)-dependent coenzyme Q10 oxidoreductase. Coenzyme Q10 is a vitamin-like compound, an endogenously produced isoprenyl benzoquinone, naturally occurring in the human body at the highest concentrations in the heart, liver, kidneys, and pancreas [7]. Coenzyme Q10 is required in the mitochondrial electron transport chain and lysosomal membranes for energy production, and it can be used as a lipophilic free radical scavenger [7]. Scientific research suggests that overexpression of FSP1 significantly inhibits the peroxidation of certain polyunsaturated fatty acids (PUFAs) and ferroptosis induction by RSL-3, a GPX4 inhibitor, and positively correlates with resistance to ferroptosis in various cancer cell lines. Further investigations into the antioxidant capabilities of coenzyme Q10 may yield benefits in mitigating ferroptosis-related damage [7].
Tetrahydrobiopterin (BH4) is a component of the antioxidant system, and it participates in nitric oxide metabolism, neurotransmitters, and aromatic amino acids. GTP cyclohydrolase-1 (GCH-1) is an enzyme that limits the BH4 synthesis rate [8]. Recent scientific literature reports show that GCH1 overexpression significantly inhibits the peroxidation of some polyunsaturated fatty acids (PUFAs) and ferroptosis induction by RSL-3, a GPX4 inhibitor [4, 5] – summary in Figure 2.

Regulators of ferroptosis
Since the discovery of ferroptosis, many researchers have attempted to identify various ways to activate this process. Currently, various small-molecule drugs are known to initiate ferroptosis in 4 ways: the first class consumes intracellular GSH; the second class targets GPX4 and deactivates its activity; the third class consumes GPX4 and the endogenous antioxidant CoQ10; and the fourth class induces lipid peroxidation by increasing the LIP pool or iron oxidation. However, there are also small-molecule compounds whose mechanism of action is more complicated and does not fit into any of the above categories [9]. Table II provides examples of ferroptosis inducers categorized by class.
Table II
Ferroptosis inducers based on scientific literature [2, 9, 21, 24]
One of the best-known inducers of ferroptosis is erastin. This compound was originally discovered in 2003 by Sonam Dolma and her research team [9, 10]. Their research aimed to explore the effects of killing different compounds on cancer cells through screening experiments. Erastin was then identified as a small molecule capable of selectively killing cancer cells expressing the oncogenes ST and RASV12 [10]. Later, erastin was extensively studied as an inducer of ferroptosis [9].
Erastin can interact with various molecules, such as the cystine-glutamate transport receptor (system Xc–), the voltage-dependent anion channel (VDAC), and the p53 protein, to trigger reactions that lead to lipid peroxidation and ultimately cell death via ferroptosis [11].
Most ferroptosis inhibitors can be categorized into lipophilic free radical scavengers, iron chelators, and deuterated polyunsaturated fatty acid phospholipids. They have been approved for the prevention of ferroptosis and regulation of normal intracellular iron metabolism, increasing GSH levels, activating GPX4, or directly inhibiting lipid peroxidation [11]. Table III provides examples of ferroptosis inhibitors categorized by class.
Table III
Ferroptosis inhibitors based on scientific literature [2, 11, 21, 24]
Genome editing with the CRISPR/Cas-9 system
Genome editing is a type of genetic engineering method in which DNA is intentionally inserted, deleted, or modified in living cells. Precise modification of specific gene sequences of interest is considered a standard approach for a deep understanding of gene function. Targeted gene modification also has therapeutic potential [12]. In recent years, genome editing using the CRISPR/Cas-9 technique has gained immense popularity, and in 2020 Emmanuelle Charpentier and Jennifer A. Doudna were awarded the Nobel Prize in Chemistry for their work on it [13].
Components of the CRISPR/Cas-9
The name CRISPR (Clustered Regularly Interspaced Short Palindromic Repeat) refers to a unique organization of short, partially repetitive DNA sequences found in the genomes of prokaryotes. The CRISPR/Cas-9 system uses a natural method of adaptive immunity found in bacteria to defend against viruses or bacteriophages [14].
In 1987, Japanese scientist Yoshizumi Ishino and his research team accidentally discovered unusual repetitive DNA sequences in Escherichia coli while analysing the alkaline phosphatase gene. However, they did not determine the biological function of these sequences at that time [14]. In 2007, it was experimentally confirmed that CRISPR is a key element of the adaptive immunity system in prokaryotes against viruses. During the adaptation process, bacterial cells develop immunity by inserting short fragments of viral DNA into their genome. These insertion sites serve as “genetic memory” of previous viral infections [14].
The CRISPR/Cas-9 system consists of 2 subunits: the guide RNA (gRNA) and the DNA endonuclease Cas-9. The Cas-9 nuclease is responsible for cleaving the target DNA to create a double-strand break in the DNA [14]. The guide RNA (gRNA) consists of 2 parts: CRISPR RNA (crRNA) and trans-activating RNA (tracrRNA). The crRNA is approximately 18–20 base pairs long and is responsible for recognizing and binding to a specific target DNA sequence. This target sequence is typically located near the site intended for genetic editing. CrRNA is designed to have a sequence complementary to the target DNA sequence. This means that crRNA can form a stable connection with the target DNA through complementary base pairs. Thanks to this crRNA-Cas-9 complex, it can recognize and precisely bind to the target sequence within the genome [13, 14]. The role of tracrRNA is mainly to act as a structural carrier for the Cas-9 endonuclease. It is longer and contains loop sequences that are necessary for forming a complex with Cas-9. TracrRNA binds to Cas-9 and allows it to associate with crRNA, creating the crRNA-tracrRNA-Cas-9 complex [13, 14].
The crRNA-tracrRNA-Cas-9 complex is crucial for precise genome editing. TracrRNA acts as a specific structural platform on which crRNA and Cas-9 can interact. It is worth noting that the latest versions of gRNA, such as single-guide RNA (sgRNA), are synthetically designed by combining the sequences of crRNA and tracrRNA into a single molecule. In the case of sgRNA, the role of tracrRNA is absorbed by the crRNA sequences, and sgRNA serves both structural and directional functions, allowing precise targeting of the CRISPR/Cas-9 system to the desired DNA sequences [13–15].
Mechanism of action
The mechanism of CRISPR/Cas-9 genome editing can be divided into 3 steps: recognition, cleavage, and repair [13–15]. Designed sgRNA guides Cas-9 and recognizes the target sequence within the gene of interest through its 5' crRNA component, which is complementary to the target DNA pair. The Cas-9 endonuclease performs double-strand breaks at a location typically 3 base pairs away from the PAM sequence [13–15]. The PAM sequence (Protospacer Adjacent Motif) is a short DNA sequence (approximately 2–6 base pairs) located downstream from the cleavage site, and its size varies depending on the bacterial species [13, 14]. The most commonly used nuclease in genome editing tools, Cas-9, recognizes the PAM sequence 5'-NGG-3' (where N can be any nucleotide). When Cas-9 finds the target site with the appropriate PAM fragment, local DNA unwinding occurs, resulting in the formation of an RNA-DNA hybrid [13, 14, 16]. In the next step, the Cas-9 endonuclease is activated to perform double-strand DNA cleavage. The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the non-complementary strand of the target DNA, primarily resulting in double-strand breaks with blunt ends. Finally, the double-strand DNA break is repaired by the host cell’s repair mechanism [13, 14, 16]. Below is a diagram of the CRISPR/Cas-9 system with double-strand DNA break repair mechanisms (Figure 3).
Figure 3
Mechanism of the CRISPR/Cas-9 editing system. SgRNA guides the Cas-9 nuclease to specific genomic sequences. Subsequently, the Cas-9 nuclease introduces double-strand breaks (DSB) near the PAM sequences. Double-strand breaks in DNA can be repaired through 2 repair pathways: non-homologous end joining (NHEJ) and homology-directed repair (HDR). NHEJ leads to small random insertions or deletions (indels) in gene knockout experiments. HDR results in precise genome modification at the site of double-strand breaks in gene knock-in experiments using a homologous donor template (based on [13, 14]). Created using Biorender.com
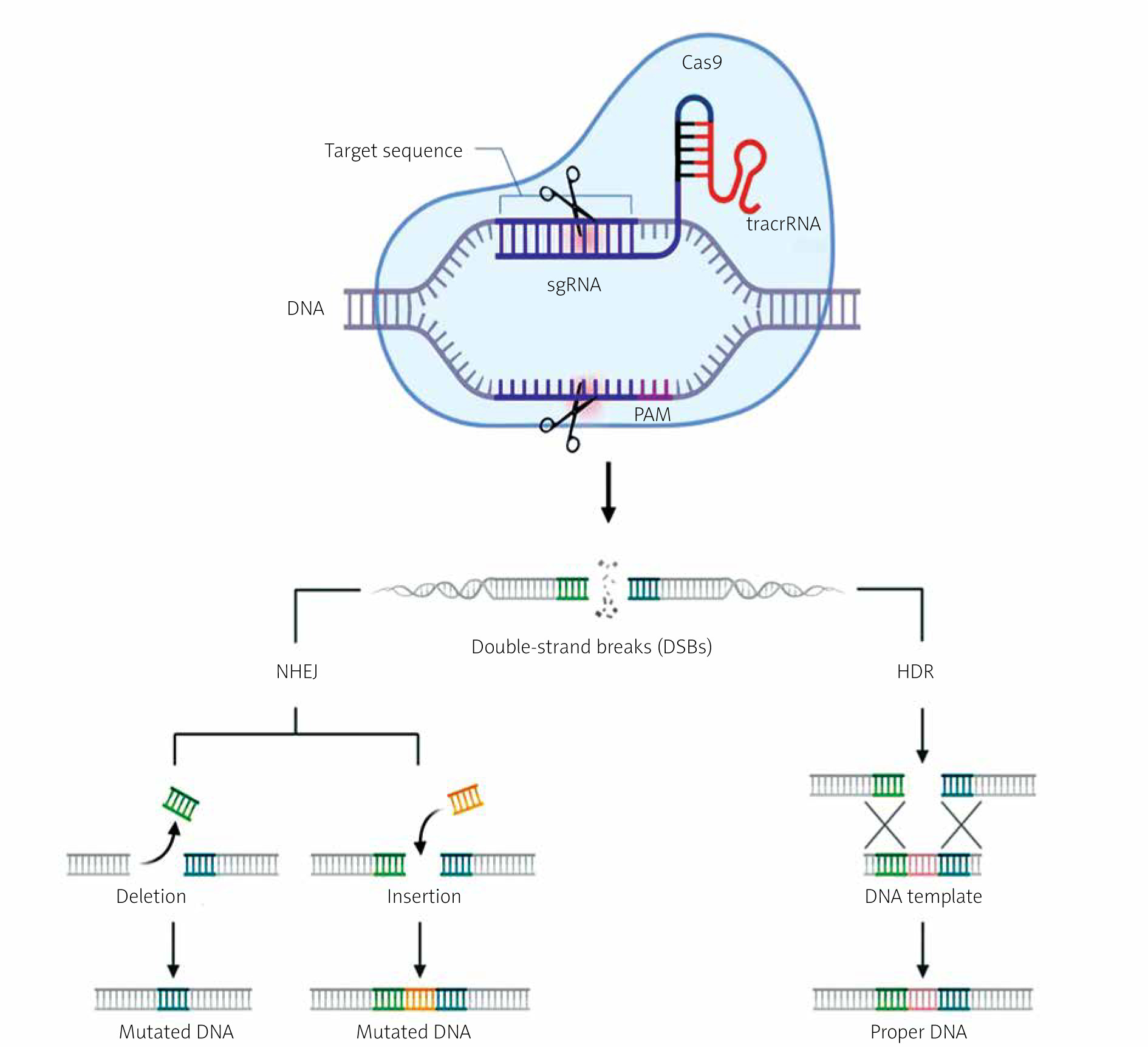
The CRISPR/Cas-9 method utilizes 2 main mechanisms for repairing double-strand breaks in DNA: non-homologous end joining (NHEJ) and homology-directed repair (HDR) [13, 14].
The non-homologous end joining (NHEJ) repair mechanism functions without the need for an external DNA template. In the case of DSBs caused by the Cas-9 protein, NHEJ can ligate the ends of the broken DNA, sometimes leading to small insertions or deletions (indels) at the repair site. NHEJ is active in all phases of the cell cycle and is relatively rapid but can result in frame-shift mutations or other imprecise DNA sequence changes [13, 14].
Homology-directed repair (HDR) uses an external DNA template with a sequence homologous to the site of the DSB. In the context of CRISPR/Cas-9, HDR can be employed to precisely insert or replace a specific DNA sequence. To achieve this, a specially designed DNA template is introduced, containing the target sequence to serve as a blueprint for repair. HDR is primarily active in the late S phase and G2 phase of the cell cycle when the necessary enzymes and replication machinery are available [13, 14]. The choice between NHEJ and HDR depends on the goal of the CRISPR/Cas-9 experiment or application. NHEJ is faster and more common but less precise, while HDR is more precise but may be less efficient in some cells [13, 14].
Oxidative stress in ferroptosis induction
The Fenton reaction (1) is an inorganic chemical reaction in which hydrogen peroxide (H2O2) and iron (Fe2+) react to produce hydroxyl radicals (•OH). In the presence of an excess of iron ions, the Fenton reaction can generate radicals that react with lipids in cell membranes, causing their peroxidation (Figure 4). This, in turn, leads to cell membrane damage and the accumulation of lipid radicals, which are characteristic of ferroptotic cell death [17]:
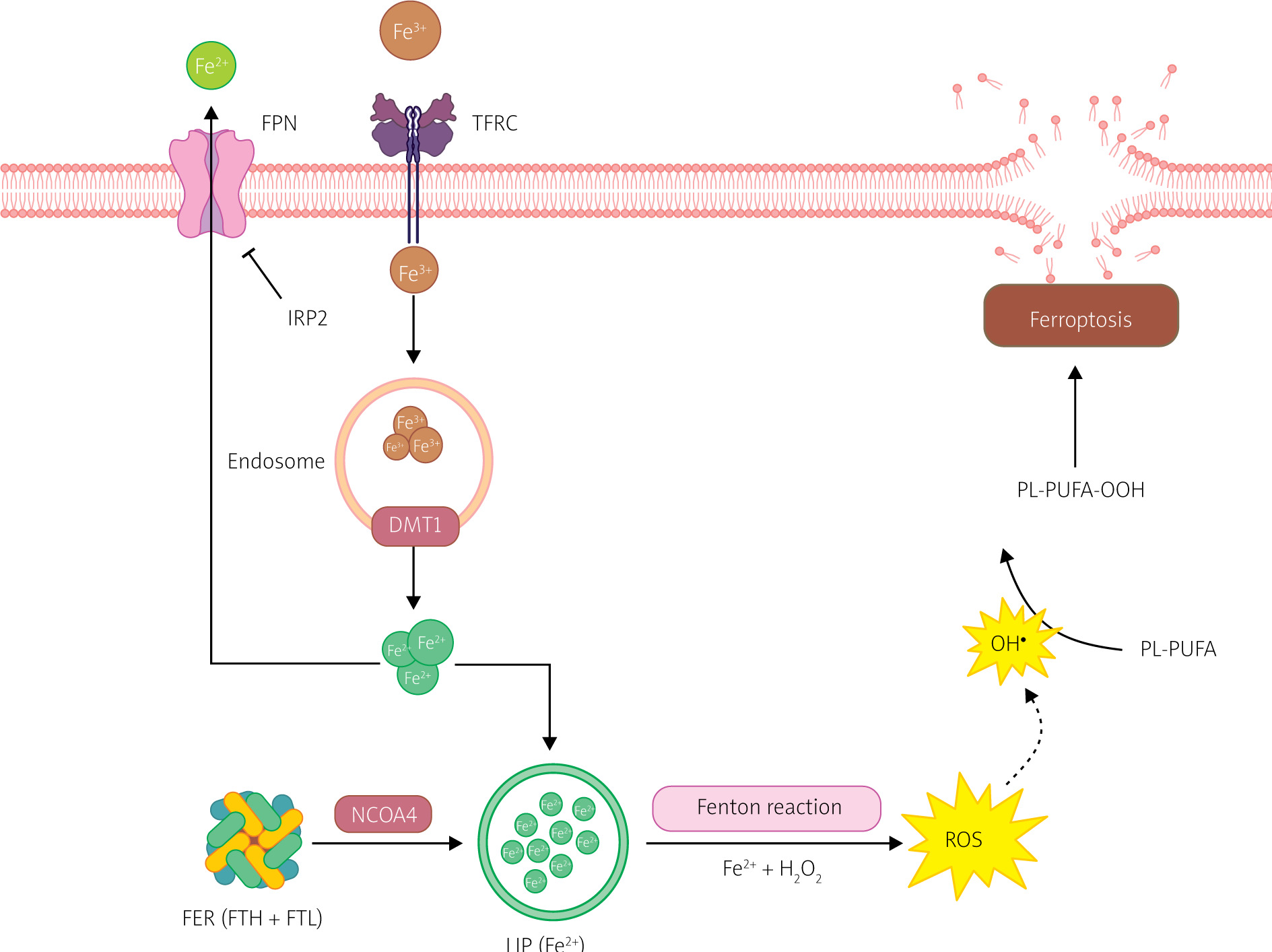
Figure 4
Iron metabolism in ferroptosis pathway. Ferroptosis is regulated by the iron metabolism pathway, which includes its absorption, transport, storage, and utilization. At the cellular level, non-heme iron is transported into cells through the transferrin receptor (TFR) relay protein. Divalent metal transporter 1 (DMT1) is responsible for releasing iron ions from endosomes, increasing the level of free iron in the cell. Additionally, heme degradation and NCOA4-dependent ferritinophagy can also increase the labile iron pool (LIP), making cells more susceptible to ferroptosis via the Fenton reaction. Created with Biorender.com
H2O2 + Fe2+ → OH• + OH– + Fe3+ (1).
Extracellular Fe3+ uptake primarily occurs through transferrin (TF) and transferrin receptor (TFRC). TF containing Fe3+ binds to TFRC, transporting Fe3+ into the cell, where it is reduced to Fe2+ [9]. Most intracellular iron is stored in ferritin (FER), with a small fraction present in a free state, forming a labile iron pool (LIP) [9, 17]. FER is a polymer consisting of heavy ferritin chain (FTH) and light ferritin chain (FTL), and it protects cells from oxidative stress caused by excess free iron [17]. The labile iron pool (LIP) is generated during FTH degradation to Fe2+, mediated by nuclear receptor coactivator 4 (NCOA4) [9, 18].
Iron regulatory proteins (IRPs), including IRP1 and IRP2, regulate intracellular iron homeostasis. IRPs bind to iron response elements (IREs) in the 5’ UTR of transcripts of iron metabolism-related genes to regulate their translation. When intracellular free iron levels are low, IRPs bind to IREs in the transcripts of FER and TFR genes, blocking FER translation and enhancing TFR translation, resulting in increased release of free iron, reduced iron efflux, and increased iron absorption [18, 19].
Ferroptosis and anticancer therapies
Despite significant breakthroughs in its therapy, cancer remains the second leading cause of death worldwide [20]. Currently, the primary therapeutic approach in chemotherapy is the use of anti-cancer drugs to induce apoptotic cell death in cancer cells. However, due to intrinsic and acquired drug resistance in cancer cells towards apoptosis, the therapeutic effect is limited. Drug resistance remains a major limiting factor in curing cancer patients [20]. Inducing ferroptosis in cancer cells is one of the promising ways to address drug resistance. There are several ways to achieve this using exogenous molecules or drugs, or by regulating physiological conditions in the external environment (e.g. high extracellular glutamine concentration), thereby blocking the Xc system [21].
Another form of anti-cancer therapy is radiotherapy, which uses ionizing radiation (IR) from a radioactive source to induce DNA damage and trigger apoptosis in cells [22]. Scientific studies report that ferroptosis inducers (such as RSL3, erastin, sorafenib, and sulfasalazine) can enhance the efficacy of radiotherapy by inhibiting SLC7A11 or inactivating GPX4 in cancer models such as glioblastoma, lung cancer, fibrosarcoma, melanoma, breast cancer, and cervical cancer [21–23]. In addition to reducing the level of SLC7A11, radiotherapy also increases ACSL4 levels, thereby increasing lipid synthesis and peroxidation, ultimately resulting in ferroptosis induction [21].
Immunotherapy is currently one of the most promising methods for treating cancer. It is based on mobilizing the patient’s immune system to fight cancer [21–23]. In recent years, it has been found that ferroptosis is closely related to immunoregulation. For example, programmed death receptor-1 (PD-1) antibodies can promote lipid peroxidation-dependent ferroptosis in cancer cells, while CD8+ T cells can decrease the expression of SLC3A2 and SLC7A11, reducing cystine uptake and promoting lipid peroxidation in cancer cells [21–23]. Transforming growth factor β (TGF-β1) can promote ferroptosis by inhibiting SLC7A11 transcription, and its inhibitors and PD-1 antibodies can synergistically generate an immunogenic tumour microenvironment and produce H2O2, thereby promoting the Fenton reaction, inducing ferroptosis in cancer cells [24].
Summary
Under certain circumstances, ferroptosis may have pro-tumour progression or anticancer effects. Released from tumour microenvironment exosomes, damage-related molecular patterns (DAMPs) lead to macrophage polarization into the M2 phenotype, which stimulates further tumour growth [24]. Despite promising prospects, the use of ferroptosis mechanisms described in this review in cancer therapy remains at the stage of experimental preclinical research. Further research is necessary to better understand these mechanisms and develop effective and safe therapies based on ferroptosis pathway modifications. Colorectal cancer is one of the most common causes of cancer-related deaths worldwide, and its progression is associated with a set of genetic changes [25]. CRISPR/Cas9 and the single guide RNA (sgRNA) system have been used to construct gene-editing tools aimed at editing mutations in genes and restore their normal biological functions. Repairing the mutation could slow down the proliferation of cancer cells, and it is a new method of gene therapy in cancers [26]. Other studies using the CRISPR/Cas9 method are still in the preclinical testing phase [25, 27]. The article presents the use of the CISPR/Cas9 method for ferroptosis, while scientists also use this method in research related to autophagy [28] in the induction of pyroptosis [29] or necroptosis [30] to be able to use the method to induce a selected type of death in supportive therapies in the future.