

Introduction
22q11.2 deletion syndrome (22q11.2DS) is known to be the most common chromosomal microdeletion disorder, with an estimated prevalence of 1 : 4000 live births [1, 2]. This syndrome is primarily caused by hemizygous microdeletion of 2.5 million base pairs (Mb) between low copy repeats (LCRs) located on the long arm of chromosome 22. The most typical manifestation is named DiGeorge syndrome, first described by Angelo DiGeorge in 1965, which is characterized by the triad of cardiac outflow tract defects, thymic hypoplasia and hypocalcemia (due to parathyroid hypoplasia). There are highly variable phenotypes in 22q11.2DS ranging from congenital anomalies (such as craniofacial features, cardiac malformation, palatal defects, parathyroid insufficiency) to later-onset conditions (including organ abnormality, development delay, immune and endocrine dysregulation, malignancy, neurological disorders and psychiatric problems) [3, 4]. Autoimmunity is suggested to have a high frequency in 22q11.2DS due to impaired thymic development [3]. Here we present a case of a 22q11.2DS patient who was diagnosed with systemic lupus erythematosus (SLE).
Case report
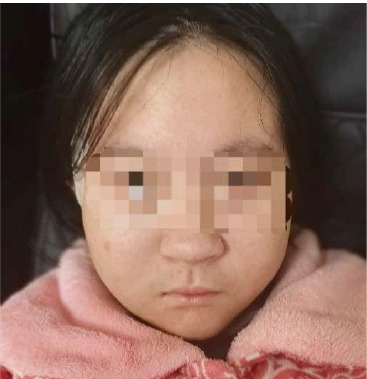
A 12-year-old girl presented to our hospital with thrombocytopenia, which was incidentally discovered during an outpatient visit. The patient was delivered by cesarean section at 35 weeks due to maternal oligohydramnios, with a birth weight of 1400 g (below the 3rd centile) and length of 41 cm (below the 3rd centile). The occipital-frontal circumference (OFC) was not measured. Prenatal ultrasound at 34 weeks gestation revealed the abdominal circumference (AC) of 246 mm (below the 3rd cen- tile) and an estimated fetal weight (EFW) of 1352 g (below the 3rd centile), indicating fetal growth restriction. Her mother denied consanguineous marriage, familial history of genetic or autoimmune disorders, and exposure to teratogens during pregnancy. The patient was an only child. She had normal motor development but exhibited short stature from an early age. Three years ago, the patient was diagnosed with growth hormone deficiency and began treatment with recombinant human growth hormone (rhGH) 2.5 IU per day. Two years ago, a chromosome microarray was performed due to intellectual developmental delay and poor academic performance, revealing a 1.5 Mb microdeletion on 22q11.2 (46,XX,arr [hg19] 22q11.21q11.22(21465661-22962196)×1). The deletion spanned LCR22D-E and was indicative of a distal type I microdeletion. Her parents were advised to undergo fluorescence in situ hybridization (FISH) analysis for the deletion region as well, but they refused. Meanwhile, she underwent assessment using the Wechsler Intelligence Scale for Children – Revised (WISC-R), indicating a below-average intelligence quotient (IQ) score of 73. The OFC measured 47 cm, suggesting microcephaly. The geneticist identified dysmorphic features including auricular anomalies (cupped ears) and micrognathia (Fig. 1). One year ago the patient underwent a liver biopsy in the Department of Hepatology in our hospital due to two-month liver dysfunction, which confirmed nonalcoholic fatty liver. Subsequently, she was diagnosed with insulin resistance as well and began treatment with metformin 0.5 g twice a day.
We inquired about her medical history in detail. She complained of persistent alopecia, intermittent mouth ulcers and low-grade fever over the past six months. However, she did not seek medical attention for these symptoms. Upon admission, her body temperature rose to approximately 37.6°C every 2-3 days and spontaneously dropped. She denied any history of arthralgia, dryness of mouth or Raynaud’s phenomenon. No skin rash or purpura was observed.
Laboratory tests revealed a decreased platelet count of 42 × 109/l, while the white blood count and hemoglobin level were normal (Table 1). The acute-phase reactant erythrocyte sedimentation rate (ESR) was elevated to 32 mm/h, but the C-reactive protein (CRP) was within normal limits. The urinalysis was unremarkable. The anti- nuclear antibody (ANA) was positive at a titer of 1 : 100 and multiple autoimmunity antibodies were detected, including anti-double strand DNA(ds-DNA), anti-riboso- mal P protein antibody (anti-P), anti-nucleosome antibody (ANuA), perinuclear anti-neutrophil cytoplasmic antibody (pANCA) and anti-glomerular basement membrane antibody (anti-GBM). Paradoxically, myeloperoxidase was negative. Light chain kappa and lambda were slightly elevated to 14.8 g/l (normal reference [NR] 6.29-13.50) and 7.9 g/l (NR 3.13-7.23), respectively. Immunoglobulin and complement were normal. The computed tomography scan showed paraseptal emphysema and reduced liver parenchyma density, while B-ultrasonography indicated accessory spleen. The electrocardiogram and echocardiogram showed no obvious abnormality. Bone marrow biopsy did not demonstrate any hematological malignancy.
Table 1
Results of laboratory tests after admission
The patient presented with alopecia, oral ulcers, low-grade fever and a decreased platelet count, along with positive ANA and ds-DNA test results, which led to a diagnosis of SLE. After excluding underlying infections and tumors, we initiated treatment with prednisone 10 mg per day and hydroxychloroquine 0.1 g twice a day. After the initiation, there was no recurrence of fever. A reevaluation after three days demonstrated an elevation in platelet count to 101 × 109/l and a reduction in ESR to 29 mm/h. We subsequently added azathioprine 50 mg per day as a steroid-sparing agent. She was discharged after five days.
We have followed up the patient until now. The aforementioned symptoms above have been alleviated. The platelet count remained within a normal range as well. Currently, her prednisone dose has been reduced to 2.5 mg per day, while maintaining the original dose of oral hydroxychloroquine and azathioprine. Unfortunately, one year after discharge, the patient was diagnosed with severe depression and mild anxiety, leading to dropping out of school. Despite the successful management of the lupus condition, her mental health has encountered new challenges.
Discussion
We present a case report of a teenager with 22q11.2 deletion who was ultimately diagnosed with SLE. She presented with low fever, mouth ulcers, alopecia, thrombocytopenia and multiple positive antiantibodies. Her clinical features were consistent with 2019 ACR/EULAR classification criteria for systemic lupus erythematosus. Moreover, previous medical records revealed that she had short stature with growth hormone deficiency, low intelligence, microcephaly, dysmorphic features, nonalcoholic fatty liver and abnormal glucose tolerance with insulin resistance treated with metformin. Imaging examinations showed the existence of an accessory spleen and paraseptal emphysema. Afterwards, she received a diagnosis of severe depression, accompanied by mild anxiety.
It has been estimated that up to 75% of pediatric 22q11.2DS patients exhibit immunodeficiency caused by thymic dysplasia. Immunodeficiency may manifest in diverse forms, including recurrent infections, poor vaccine responses, autoimmune disorders and atopy. The dysfunction of thymus directly leads to peripheral T-cell lymphopenia in early infancy [5]. The majority of cases present as mild and usually occur in childhood, which can be gradually corrected with age as there is a homeostatic expansion mechanism [5, 6]. The maturation and function of B lymphocyte are also disturbed secondary to cellular immunocompromise, encompassing immunoglobulin A (IgA) deficiency, hypo- gammaglobulinemia, defects in functional antibody production and low switched memory B cells [4, 7]. Complement activation has been observed, indicated by increased plasma levels of C3bc and terminal complement complex [2]. There is also a decrease in the number of circulating dendritic cells, affecting both myeloid and plasmacytoid subsets [8].
The overall frequency of autoimmunity in 22q11.2DS is estimated at approximately 10% [9, 10]. Recent larger-scale cohorts and longer-term follow-up studies have revealed a notable increase in prevalence, ranging from 18% to 29.6% over the past decade [11-14]. The potential mechanism is suggested to be chronic infections, deficiency of regulatory T cells, defective thymic central tolerance and decreased repertoire diversity [15]. Higher percentages of memory T-helper cells and inflammatory Th1 and Th17 cells result in the promotion of an inflammatory state [16]. The reduction of anti-inflammatory interleukin (IL)-10 and the increase of IL-17, IL-6 and IL-23 are observed in 22q11.2DS [17]. Abnormal specific antibodies may also be attributed to the reduction of memory B cells and plasmacytoid dendritic cells [8, 18]. Arthritis, cytopenia, dermatosis, endocrinopathies, vasculitis, enteropathy and encephalitis with autoimmune features have been previously reported [10, 19], among which immune thrombocytopenia, hemolytic anemia, thyroiditis and arthritis are most commonly described. Other sporadic cases encompass conditions such as psoriasis, vitiligo, Raynaud’s phenomenon and inflammatory bowel disease (IBD). A multicenter study conducted by Montin et al. [13] revealed that the prevalence of hematological autoimmunity was 8%. As Ricci et al. reported, 21.9% of the total group developed autoimmune thyroid diseases, with half of them ultimately requiring treatment [20]. The prevalence of juvenile rheumatoid arthritis was identified as 3.75% by Sullivan et al. [21]. The decrease in specific subpopulations of T lymphocytes can potentially serve as a predictive factor for autoimmunity [22, 23]. It is recommended to conduct systemic immunophenotyping at diagnosis to identify high-risk autoimmune conditions early in 22q11.2DS [13].
Systemic lupus erythematosus is rare in patients with 22q11.2DS. We searched the literature in PubMed and found only one article. Lin et al. reported a 22-year-old SLE patient who was identified to have central 22q11.2 DS. She suffered from cutaneous rash, antiphospholipid syndrome, nephritis and hemocytopenia [18]. Her positive antibodies included ANA, ds-DNA, anti-P and Sjogren’s syndrome-related antigen A (SSA). The genetic report showed a ~737 kb heterozygous deletion within LCR22B-D (chr22: 20,728,956-21,465,659).
In contrast to the above, our patient’s 1.5 Mb deletion was located within LCR22D-E (chr22: 21,465,661-22,962,196), involving 13 OMIM genes, including GGT2, HIC2 and MAPK1. This region is classified into the distal type I deletion group. The major clinal manifestations of this subtype include preterm birth, growth restriction, cardiac defects, dysmorphic features, minor skeletal anomalies, microcephaly and developmental delay [24], but there are a few reports linking it to autoimmune diseases.
It is noteworthy that this patient exhibited multiple endocrine and metabolic disorders, including growth hormone deficiency, impaired glucose tolerance and non-alcoholic fatty liver. According to a previous study, the frequency of growth hormone deficiency in 22q11.2DS is approximately 4% [25], while Lin et al. reported a higher prevalence of short stature of up to 40% [26]. More than one-third of patients are below the third centile for either height or weight parameters [27]. Weinzimer recommended routine monitoring of growth speed and performing necessary tests for hormone levels when a significant slowdown occurs [25]. Type 2 diabetes (T2D) affects about 5% of individuals with 22q11.2DS and is diagnosed at an earlier age [28, 29]. Van et al. identified 22q11.2DS as an independent risk factor for early-onset T2D [30]. Soubry et al. found that 28% (21 of 75) of the patients with 22q11.2DS had liver steatosis [29]. These findings provide a possible explanation for our observation in the young patient, who is not taking antipsychotic drugs and does not have obesity.
Furthermore, the patient had an accessory spleen and paraseptal emphysema. While there have been occasional reports of accessory spleens in patients with 22q11.2 DS [31-34], no data are available to establish their prevalence. Our literature search did not find any evidence linking paraseptal emphysema with 22q11.2DS. We speculate that it may be associated with autoimmune features [35].
At least 73-90% of patients with 22q11.2DS were diagnosed with psychiatric diseases [36]. A large-scale collaborative study found that the prevalence of anxiety was 40-76%, and the prevalence of mood disorders was 9-35% [37]. These results underscore the significance of ongoing monitoring and efforts to alleviate the enduring psychopathological burden in individuals diagnosed with 22q11.2 DS.
Conclusions
22q11.2 deletion syndrome is a common microdeletion syndrome that has been associated with autoimmune diseases. Our report adds to the limited literature on the rare co-occurrence of 22q11.2DS and SLE. This case underscores the importance of considering genetic factors, such as 22q11.2DS, in the diagnosis of rheumatic diseases that manifest in childhood. Early diagnosis and treatment of SLE in patients with 22q11.2DS may lead to better outcomes and an improved quality of life.