






Introduction
One of the main reasons of death in all countries is cancer, causing around 10 million deaths were recorded in 2020 [1]. According to the World Health Organization (WHO), the main frequently encountered reason for cancer deaths in 2020 was lung cancer (1.80 million deaths) [2]. Non-small cell lung cancer (NSCLC) is the most common form of lung cancer, accounting for around 85% of lung cancers. Cisplatin combination chemotherapy is currently used as a standard treatment for patients having NSCLC. The survival rate ranges from approximately 6.9–11.3 months, and the response rate of patients to chemotherapy ranges 20–35%. Patients suffering from lung cancer often develop resistance to cisplatin. Drug resistance to chemotherapeutic drugs remains one of the main reasons for the failure of several cancer treatments such as lung cancers [3]. Recently, medicinal plants are considered as one of the most efficient and safest medical therapies. These herbal treatments are also the main source of flavonoids, which play an important role in the healing and management of cancer. Additionally, the fact that these medications generally have fewer side effects than synthetic medications has led researchers to investigate their molecular mechanisms [4]. According to the WHO, herbal medicine accounts for around 80% of the health care in most developing countries. Many studies have declared that herbal treatment may inhibit tumour proliferation and induce cytotoxic properties in malignant cells without causing any side effects in normal cells. Interestingly, the apoptosis pathway is one of the most essential signalling pathways which can be modulated by anticancer agents. Many studies have suggested that extracts derived from plants may play a role in cancer treatment and prevention [5].
Hesperidin, a flavonoid glycoside [6], has been demonstrated to have anticancer properties by increasing apoptosis and suppressing proliferation in numerous cancer types, including breast and lung cancer [7]. Hesperidin is 5, 7, 3-trihydroxy-4-methoxy-flavanone7 rhamnoglucoside. Hesperidin suppresses proliferation and stimulates apoptosis in lung malignant cells, without any toxic effects on normal epithelial lung cells. Additionally, hesperidin inhibits the invasion and migration of lung malignant cells by regulation of the SDF1/CXCR4 axis [8]. In in vivo studies, hesperidin pretreatment effectively protects against the progression of carcinogen-induced cancer of lungs from several carcinogens. In vitro studies have shown that hesperidin reduces the proliferation of NSCLC cells by controlling the activation of several pathways linked to apoptosis and immunological reactions. To treat NSCLC, hesperidin may be employed as a novel anti-cancer agent [9].
To promote the proliferation, cytokine production, and cytotoxicity of cancer cells, programmed cell death ligand-1 (PD-L1), a transmembrane protein, selectively binds to PD-1 [10]. According to reports, the overexpression of PD-L1 in cancer cells promotes the growth of the disease by making the cells more resistant to pro-apoptotic stimuli like interferon and by activating several transcription factors like nuclear factor κB (NF-κB) and epidermal growth factor (EGF) [11]. Nuclear factor κB induction includes both canonical and non-canonical signalling pathways, which are responsible for regulation of inflammatory responses via several mechanisms. These signalling pathways have been investigated because they are essential in the inflammatory response and have been confirmed to be linked to chronic inflammatory diseases. Therefore, there is a need to identify a natural compound that can decrease inflammatory gene expression, such as those associated with NF-kB signalling [12]. Several studies noted that these pathways have been established to play a role in the expansion, proliferation, suppression of apoptosis, angiogenesis, and invasion of cancer cells. Inflammatory enzymes and mediators, CDs, and T-cell activation markers are promising diagnostic and treatment hit candidates in various non-communicable diseases [13–17]. As a result of its multiple roles in carcinogenesis, EGF receptor (EGFR) has been viewed as a promising applicant for anticancer therapy [18, 19].
Small non-coding RNAs, known as miRNAs, control a variety of biological processes, including carcinogenesis, which is dysfunctional in some cancer cells [20–22]. Additionally, miRNAs can be efficient negative regulators of gene expression and can control the invasion, migration, and metastasis of cancer cells [23]. MiR-34a can act as a potent tumour suppressor because of its capacity to control the expression of several target oncogenes implicated in the progression of cancer [24]. Hesperidin may act as a regulator of cancer cell migration and invasion, and some studies have shown that hesperidin administration potentially increases miR-34a expression and decreases NF-κB expression [25–27].
Recent studies have focused on how hesperidin can alter the characteristics of cancer illnesses by reducing pro-inflammatory enzymes and mediators, strengthening the antioxidant defence system, and reducing the proliferation of cancer cells by enhancing the apoptotic process in cancer cells [28–30]. Given this context, the purpose of the current study is to investigate the effect of hesperidin on the apoptosis and proliferation of NSCLC cells A549 and H460 via targeting the miR 34a/PD-L1/NF-κB signalling pathway. These results may justify the utilisation of hesperidin being a therapeutic adjuvant in numerous situations to prevent medication resistance.
Material and methods
Chemicals
Hesperidin (≥ 80% purity) was procured from (Sigma- Aldrich; Merck KGaA) (St. Louis, MO, USA). Cisplatin (Cisplatin,®Mylan, 50 mg/50 ml vial) was bought from Biodiagnostics Company (Cairo, Egypt). For the preparation of solutions, hesperidin powder was dispersed in DMSO (Sigma-Aldrich; Merck KGaA) and afterward diluted to 100, 300, 1000, 3000, and 10000 µM with cisplatin, and RPMI-1640 medium was dispersed in sterile phosphate-buffered saline (PBS) to a concentration of 1 mg/ml, and then the culture medium was diluted to 0.01, 0.1, 1, 10, and 100 µM.
Cell culture
The human lung cancer cell lines A549 and H460 were bought from Nawah Scientific Inc. in Mokatam, Cairo, Egypt. Cells were kept at 37°C in DMEM media with 10% heat-deactivated foetal bovine serum, 100 mg/ml streptomycin, and 100 units/ml penicillin. The environment was humidified with 5% (v/v) CO2.
Cytotoxicity assay
Cell viability was measured using the cell viability assay – the protein dye sulforhodamine B (SRB) test, which is based on a dye binding to basic amino acids in cellular proteins. Aliquots of a 100 µl cell suspension (5 × 10–3 cells) were cultivated for 24 hours in complete medium in 96-well plates. To treat the cells, a second aliquot of 100 µl of media with various doses of medication was employed. After 48 hours of exposure to hesperidin and cisplatin, the cells were settled by eliminating the medium and changing it with 150 µl of 10% trichloroacetic acid (TCA), which was then incubated for one hour at 4°C. The cells were washed with distilled water 5 times after the TCA solution was removed. 70 µl of 0.4% w/v SRB solution was introduced to the mixture in aliquots, and it was then incubated for 10 minutes at room temperature and in the dark [31]. Plates were washed with 1% acetic acid 3 times prior to being allowed to air dry overnight. 150 µl of TRIS (10 mM) was then added to dissolve the protein-bound SRB dye. Then, using a BMG LABTECH®- FLUOstar Omega microplate reader (Ortenberg, Germany), the absorbance was calculated at 540 nm [32]. The cytotoxic activity was estimated as the IC50, which is the concentration required to decrease the absorbance of treated cells by 50% compared to the untreated cells. For IC50 determination, a dose-response curve is plotted between the drug concentration and percent growth inhibition, as calculated here for hesperidin and cisplatin in both lung cancer cell lines. IC50 values can be determined using IC50 calculation software. The percentage of cell viability is calculated using the following equation:
% viability = mean OD sample/mean OD blank × 100
where OD mean optical density.
Cell cycle assay by flow cytometry
After being exposed to hesperidin and cisplatin at IC50 concentration for 48 hours, 814.36 and 944.21 µM for hesperidin and 11.18 and 27.6 µM for cisplatin, respectively, for A549 and H460 cells, cells (105 cells) were harvested by trypsinization and cleaned twice with ice-cold PBS (pH 7.4). Suspended cells were fixed in 2 ml of 60% ice-cold ethanol for an hour at 4°C. They were then cleaned twice more with PBS (pH 7.4) before being re-immersed in 1 ml of PBS with 50 g/ml RNAase A and 10 g/ml propidium iodide (PI). After incubation for 20 min in the dark at 37°C, cells were analysed by flow cytometry utilising an FL2 (ex/em, 535/617 nm) signal detector (ACEA Novocyte TM flow cytometer, ACEA Biosciences Inc., San Diego, CA, USA). For each sample, 12,000 occurrences were gathered. Cell cycle distribution was determined using ACEA Novo Express TM software (ACEA Biosciences Inc., San Diego, CA, USA). Triplicates of experiment were performed.
Wound healing assay
For the scratch wound experiment, cells were plated at a density of 2 × 105 per well on a coated 12-well plate and cultivated overnight. The confluent monolayer was scratched the following day, and the plate was carefully cleaned with PBS. While drug wells were treated with fresh media containing the drug, control wells were supplied with fresh medium. Between each time point, the plate was incubated at 37°C and 5% CO2. Cell migration from the intact region into the scratched zone could be seen under a microscope. The percentage of wound closure was measured and compared to the value obtained at zero hours using images captured under an inverted microscope at the designated time intervals. Cell migration was suggested by the rise in the closure percentage. Using MII, the average distance between scratch edges was used to compute the wound’s breadth. The wound area was calculated by tracing the region of an image that was free of cells using Fiji-Image J software [33]. Triplicates of each experiment were performed.
Western blot technique
A549 and H460 cells that had been exposed to hesperidin for 72 hours were then treated with ice-cold RIPA lysis buffer (Beyotime Institute of Biotechnology) to extract the total protein. According to the manufacturer’s instructions, each sample of the cell pellet was mixed with the ReadyPrepTM protein extraction kit (total protein) offered by Bio-Rad Inc. (Catalogue #163-2086). The amount of protein in each group was measured using a bicinchoninic acid assay. Proteins were loaded onto a 10% SDS gel, transferred to PVDF membranes, and blocked with a 5% skimmed milk solution for 2 hours at room temperature. Human anti-actin primary antibodies were used to probe membranes (1 : 4000; cat. no. ab179467; Abcam) and rabbit anti-NF-κB (1 : 1000; ab16502; Abcam) at 4°C. Ultimately, the membranes were treated with goat anti-rabbit antibody coupled to horseradish peroxidase for a further 3 hours at room temperature (ab6721; Abcam). The next step was to visualise the signals with a chemiluminescence solution (EMD-Millipore). The experiment was performed in triplicate.
Quantitative reverse transcription polymerase chain reaction and RNA isolation
To extract the total RNA from A549 and H460 cells, a TRIzol reagent kit (Invitrogen; Thermo Fisher Scientific, Inc.) was utilised. A Thermo Fisher Scientific, Inc. NanoDrop 3000 spectrophotometer was used to measure the concentration of the extracted RNA. Takara Bio, Inc. PrimeScript RT reagent kits were used to perform reverse transcription on the isolated RNA, producing cDNA and oligo primers. For RT, the following methods were employed: 5 seconds at 85°C and 15 minutes at 38°C. The Power SYBR Green PCR Master mix (Takara Bio, Inc.) was utilised to measure the quantitative polymerase chain reaction (qPCR). The expression of the P53 mRNA levels was normalised using GAPDH as an internal control, PDL-1, EGFR, and MiR-34 in the qPCR. The primer sequences for these were as follows: P53 forward, 5’ CCTATCCGGTCAGTTGTTGGA 3’ and reverse, 5’- TTGCAGAGTGGAGGAAAT 3’; PDL-1 forward, 5’ GCCAGGGTTTTCCCAGTCACG 3’ and reverse, 5’ GAGCGGATAACAATTTCACA 3’; EGFR forward, 5’-GCCAGGGTTTTCCCAGTCACG-3’ and reverse, 5’ GAGCGGATAACAATTTCACA 3’; MiR-34 forward, 5’ TGGCAGTGTCTTAGCTGGTT 3’ and reverse, 5’ GAACATGTCTGCGTATCTC 3’ and GAPDH forward, 5’ TGGATTTGGACGCATTGGTC -3’ and reverse, 5’ TTTGCACTGGTACGTGTTGAT 3’. Using the 2-ΔΔCqtechnique, the relative mRNA expression of P53, PDL-1, EGFR, and MiR-34 was determined. The experiment was performed in triplicate.
Statistical analysis
Data were analysed and graphing was performed using GraphPad Prism, version 7.0 (GraphPad Software, Inc.). Statistical results are presented as the mean ± standard deviation of 3 independent experiments. Differences among several groups were compared using one-way ANOVA with Bonferroni test in cases when all groups were compared or a post-hoc Dunnett’s test in cases when all groups were compared with the control group. Student’s t-test was used to analyse the differences between 2 groups. P < 0.05 was considered a significant difference.
Results
Effect of hesperidin vs. cisplatin on the viability of A549 and H460 lung cancer cells as determined by sulforhodamine B assay
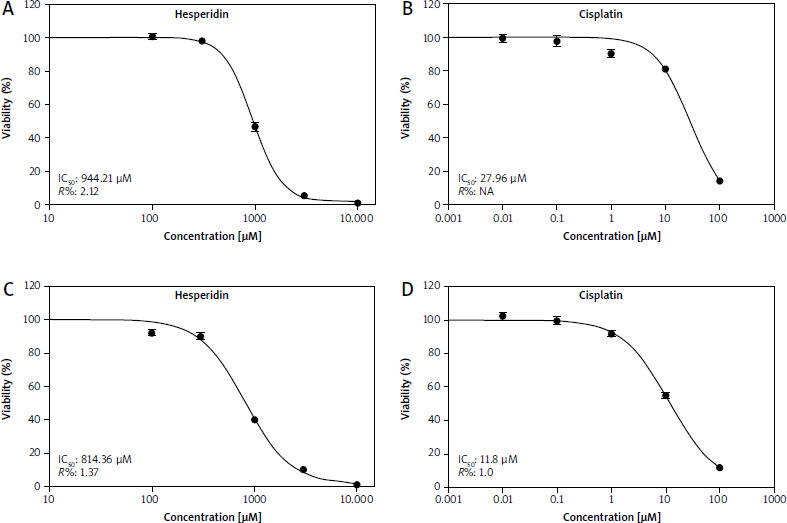
Based on the mentioned results, hesperidin produced cytotoxic effects that changed with time and concentration in these cells at increasing concentrations of 100–1000 µM when compared to cisplatin on A549 and H460 lung cancer cells. When the concentration of hesperidin was increased to 300–1000 µM, a sharply increased cytotoxic impact was noticed. On the other hand, cell lines were exposed to concentrations of cisplatin ranging 0.01–100 µM for 48 hours tested for viability using an SRB colorimetric assay [34]. Hesperidin’s IC50 value for 48 hours was calculated using cytotoxicity plots and cell proliferation, and they were determined to be 814.36 µM and 944.21 µM for A549 and H460 lung cancer cells, respectively [35] (Figs. 1 A, C). Conversely, IC50 values of cisplatin calculated for 48 hours were estimated for A549 and H460 lung carcinoma cells to be 11.18 and 27.6 µM, respectively (Figs. 1 B, D). Cytotoxic analysis demonstrated the toxic and harmful effects of both treatments in both lung cancer cell lines. Our outcomes demonstrated that the IC50 value of hesperidin showed minor cytotoxicity, in comparison with the IC50 value of cisplatin, which showed a high cytotoxic effect. Nevertheless, our results indicated that cisplatin is accompanied by the interaction between DNA molecules and cisplatin, resulting in the generation of superoxide radicals that influence several pathways including upregulation of P53, which was clearly observed in our results by PCR assay. These results are confirmed by another study, which demonstrated that cisplatin induces the death of malignant cells and leads to other organ damage due to ROS generation [36].
Fig. 1
Effect of hesperidin vs. cisplatin on the viability of A549 and H460 cells as determined by a sulforhodamine B assay at 48 hours. Effects of various concentrations of hesperidin on the viability of H460 cells (A), effects of various concentrations of cisplatin on the viability of H460 cells (B), effects of various concentrations of hesperidin on the viability of A549 cells (C), effects of various concentrations of cisplatin on the viability of A549 cells (D). Hesperidin’s IC50 values for 48 hours were calculated using cytotoxicity plots and cell proliferation, and they were determined to be 814.36 μM and 944.21 μM, respectively, for A549 and H460 lung cancer cells [35]
As opposed to Figures 1 A, C, IC50 values of cisplatin calculated for 48 hours were estimated for A549 and H460 lung carcinoma cells to be 11.18 and 27.6 μM, respectively – Figures 1 B, D. Cytotoxic analysis demonstrated the toxic and harmful effects of both treatments in both lung cancer cell lines. As our outcomes demonstrated, the IC50 value of hesperidin showed minor cytotoxicity, in difference with the IC50 value of cisplatin, which showed a high cytotoxic effect.
Hesperidin and cisplatin individually induced cell cycle arrest in A549 and H460 lung cancer cells
To understand the mechanism of cell cycle arrest, the effects of hesperidin and cisplatin at IC50 concentration for 48 hours (IC50; 814.36 and 944.21 µM for hesperidin and 11.18 and 27.6 µM for cisplatin, respectively, for A549 and H460 cells) on the cell cycle were evaluated by flow cytometry. According to the study’s findings, which are shown in Figures 2 and 3, hesperidin caused cell cycle arrest at the Sub-G1 phase, which corresponds to the non-proliferating phase of A549 cells at frequencies of 37.89, 37.96, and 38.21 Hz, respectively (Figs. 2 D–F). On the other hand, cisplatin treatment enhanced the cell population in the G2 phase at a frequency of 70.39, 73.24, and 72.88 Hz, respectively, compared to the control A549 cells in the G2 phase (25.83, 28.15, and 20 Hz, respectively) (Fig. 2 G–I). Additionally, hesperidin administration significantly triggered cell cycle arrest at the G2 phase in H460 cells (31.65, 33.98, and 32.29 Hz respectively, in the G2 phase) (Fig. 3 A–C). The cell replication phase is indicated by G2 phase. It is anticipated that substances that interfere with microtubular spindles will greatly boost this cell population [37]. As opposed to this, cisplatin caused cell cycle arrest in H460 at frequencies of (40.37, 40.71, and 38.42 Hz, respectively) (Fig. 3 G–I). Thus, hesperidin and cisplatin treatments boosted H460 and A549 cell death by prompting apoptosis and cell cycle arrest.
Fig. 2
Hesperidin and cisplatin individually induce cell cycle arrest in A549 cell lines. Cell cycle analysis for control cells (A–C), cell cycle analysis for hesperidin-treated A549 cells; hesperidin induces cell cycle arrest in sub-G1 phase (D–F), cell cycle analysis for cisplatin-treated A549cells; cisplatin induces cell cycle arrest in G2 phase (G–I)
The experiments were performed in triplicate. Hesperidin caused cell cycle arrest at the sub-G1 phase, which corresponds to the non-proliferating phase of A549 cells at frequencies of 37.89, 37.96, and 38.21, respectively (Fig. 2 D–F). Cisplatin cure enhanced the cell population in the G2 phase at a frequency of 70.39, 73.24, and 72.88, respectively, compared to the control A549 cells in the G2 phase (25.83, 28.15, and 20, respectively) (Fig. 2 G–I). As a result, hesperidin and cisplatin treatments boosted A549 cell death by prompting cell cycle arrest.
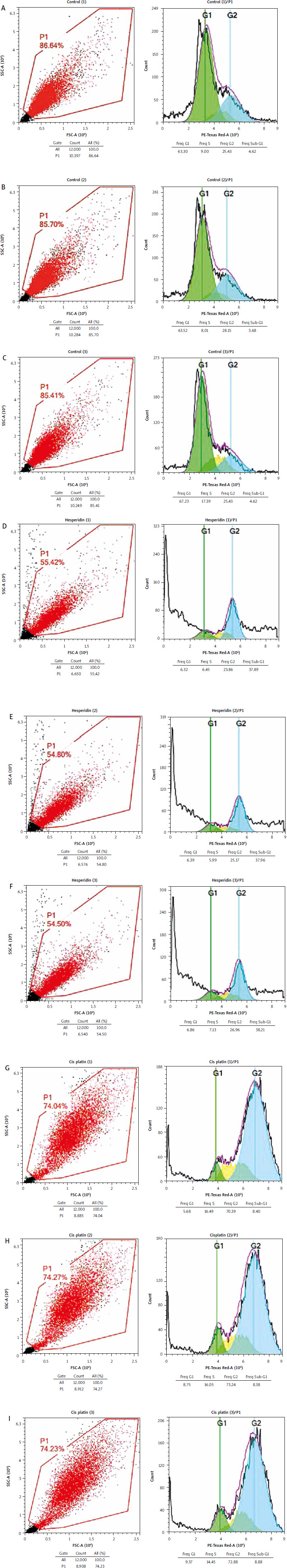
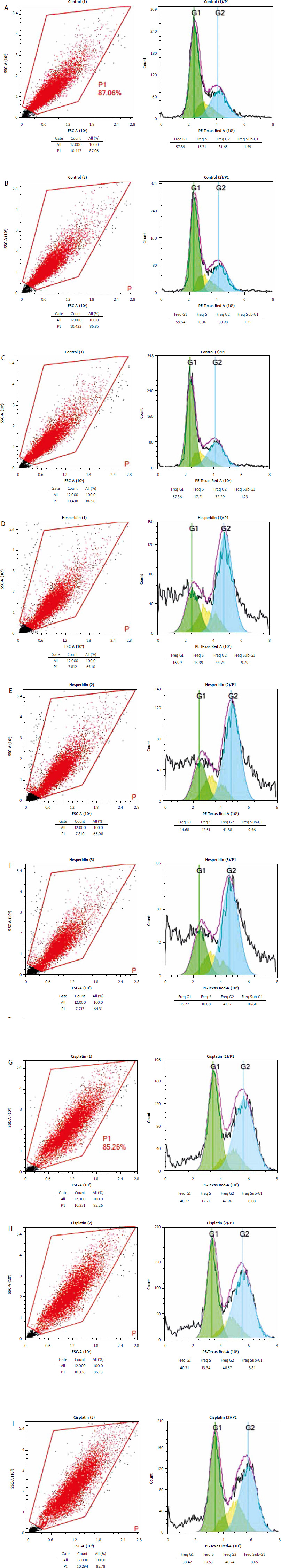
Fig. 3
Hesperidin and cisplatin individually induce cell cycle arrest in H460 cell lines. Cell cycle analysis for control cells (A–C), cell cycle analysis for hesperidin-treated H460 cell; hesperidin induces cell cycle arrest in G2 phase (D–F), cell cycle analysis for cisplatin-treated H460 cells (G–I); cisplatin induces cell cycle arrest in G1 phase
The experiments were performed in triplicate. Hesperidin administration significantly triggered cell cycle arrest at the G2 phase in H460 cells (31.65, 33.98, and 32.29, respectively, in the G2 phase) compared to untreated cells, while cisplatin caused cell cycle arrest in H460 at frequencies of 40.37, 40.71, and 38.42, respectively. As a result, hesperidin and cisplatin treatments boosted H460 cell death by prompting apoptosis and cell cycle arrest.
Hesperidin and cisplatin individually lead to inhibition of cell migration in A549 and H460 lung cancer cells
In this study, wound-healing assay was used to assess how hesperidin and cisplatin at IC50 concentration for 48 hours (IC50; 814.36 and 944.21 µM for hesperidin and 11.18 and 27.6 µM for cisplatin for A549 and H460 cells, respectively) affected the migration of A549 and H460 cells. Images of the number of cells that migrated into the scratched area were taken after treatment with hesperidin and cisplatin, the number of cells was used to estimate the migration rate. Additionally, wound width, wound closure, and migration rate were used to calculate the results of wound healing. In Figures 4 and 5 the results showed that hesperidin reduced cell migration in both cell lines in a time-dependent way. Hesperidin treatment significantly reduced the rate of cell migration in A549 and H460 cells after 48 hours from 0.015 ±0.0037 in control cells to 0.007 ±0.0041 in hesperidin treated H460 cells (Fig. 5) and from 0.04 ±0.009 in control cells to 0.02 ±0.001 in hesperidin-treated A549 cells (Fig. 4). The control group did not exhibit evidence of cell migration inhibition. Meanwhile, the treatment with cisplatin caused a significant reduction in the rate of migration of cells in both cell lines after 48 hours (Figs. 4, 5). Subsequently, when the A549 cells were treated with hesperidin (IC50), 95% wound closure was detected, and at 72 hours the wound was almost totally closed (Fig. 4). Interestingly, in hesperidin-treated H460 cells, 75% wound closure was observed at 72 hours (Fig. 5), while cisplatin-treated H460 and A549 cells showed 85% and 73% wound closure at 72 hours respectively (Figs. 4, 5). Moreover, A549 and H460 cells both treated with hesperidin and cisplatin (IC50) showed significantly higher wound with compared to untreated cells (Figs. 6, 7). The results indicated the anti-migratory potential of hesperidin and cisplatin against H460 and A549 cells.
Fig. 4
Effect of cisplatin and hesperidin on the migration of A549 cells represented by migration rate (A) wound closure as determined by a wound-healing assay (B)
The experiments were performed in triplicate at 24, 48, and 72 hours. The results are displayed as mean ± standard deviation. The results presented are means of 2 independent experiments. ap < 0.01 vs. control, bp < 0.01 vs. cisplatin-treated cells. Hesperidin treatment significantly reduced the rate of cell migration in A549 cells after 48 hours from 0.04 ±0.009 in control cells to 0.02 ±0.001 in hesperidin-treated A549 cells. When the A549 cells were treated with hesperidin (IC50), 95% wound closure was detected at 72 hours and the wound was almost totally closed. While cisplatin-treated A549 cells showed 73% wound closure at 72 hours.
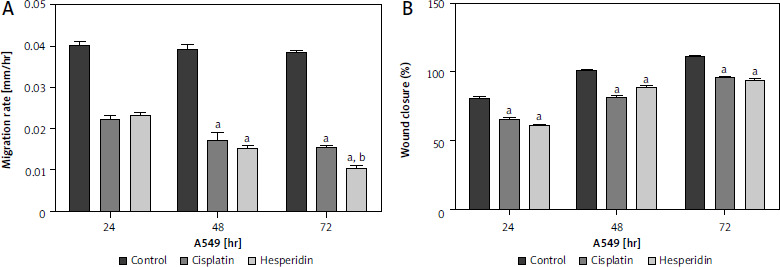
Fig. 5
Effect of cisplatin and hesperidin on the migration of H460 cells represented by migration rate (A) and wound closure as determined by a wound-healing assay (B)
The experiments were performed in triplicate at 24, 48, and 72 hours. The results are displayed as mean ± standard deviation. The results presented are means of 2 independent experiments. ap < 0.01 vs. control. Hesperidin treatment significantly reduced the rate of cell migration in H460 cells after 48 hours from 0.015 ±0.0037 in control cells to 0.007 ±0.0041 in hesperidin-treated H460 cells. The control group did not exhibit any evidence of cell migration inhibition. Meanwhile, the treatment with cisplatin caused a significant reduction in the rate of migration of cells in H460 cells after 48 hours. Interestingly, in hesperidin-treated H460 cells, 75% wound closure was observed at 72 hours, while cisplatin-treated H460 cells showed 73% wound closure at 72 hours.
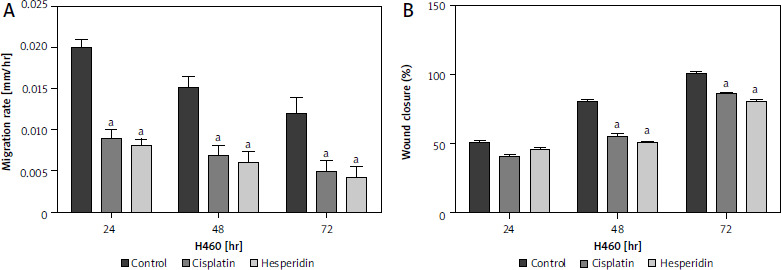
Fig. 6
Wound healing assay images for A549 cells at 72 hours. The A549 cells treated with hesperidin and cisplatin (IC50) showed significant higher wound width compared to untreated cells. Control cells (A), hesperidin-treated A549 cells (B), cisplatin-treated A549 cells, the experiments were performed in triplicate (C), statistical analysis of the wound width results (D)
The results presented are means of 2 independent experiments. ap < 0.01 vs. control. A549 cells both treated with hesperidin and cisplatin (IC50) showed significantly higher wound width compared to untreated cells. The results indicated the anti-migratory potential of hesperidin and cisplatin against A549 lung cancer cell lines cells.
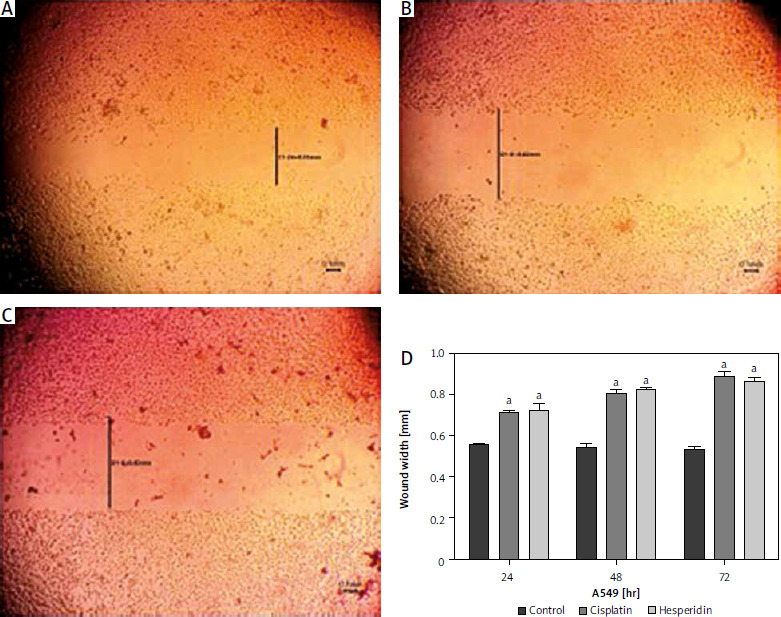
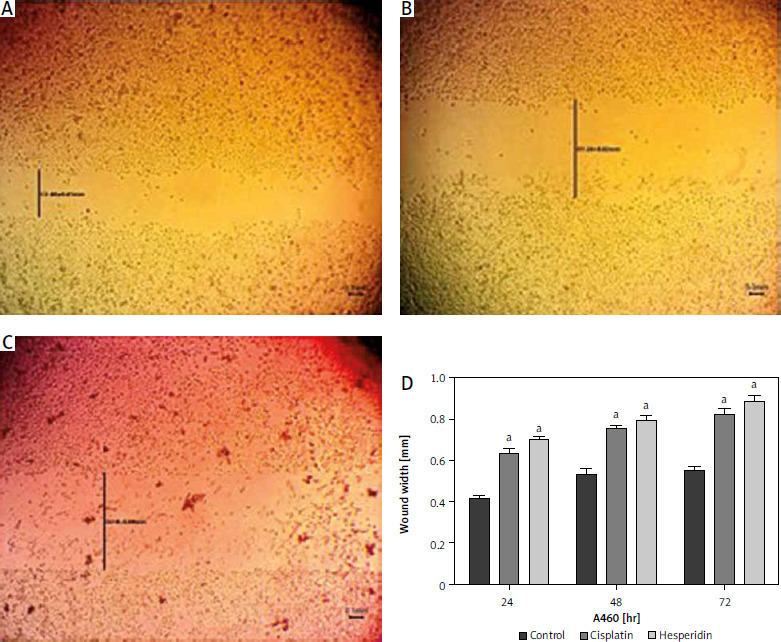
Fig. 7
Wound healing assay images for H460 cells at 72 hours. The H460 cells treated with hesperidin and cisplatin (IC50) showed significant higher wound width compared to untreated cells. Control cells (A), hesperidin-treated H460 cells (B), cisplatin-treated H460 cells, the experiments were performed in triplicate (C), statistical analysis of the wound width results (D)
The results presented are means of 2 independent experiments. ap < 0.01 vs. control. H460 cells showed significantly higher wound width compared to untreated cells when treated with hesperidin and cisplatin (IC50). The results indicated the anti-migratory potential of hesperidin and cisplatin against H460 lung cancer cell lines
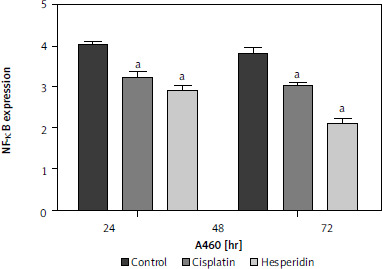
Hesperidin and cisplatin individually decreased nuclear factor κB expression in A549 and H460 lung cancer cells
To investigate the effect of hesperidin and cisplatin on NF-κB expression, NF-κB expression was detected by western blot after drug administration, as illustrated in Figure 8 – A549 and H460 lung cancer cells showed a remarkable increase in NF-κB expression. However, the administration of hesperidin and cisplatin to A549 and H460 lung cancer cells developed a notable reduction in the protein expression of NF-κB, with the lowest values detected in the hesperidin-treated H460 lung cancer cells, thereby enhancing the accretion of medicines in cancer cells and promoting the toxic influences on tumour cells.
Fig. 8
Hesperidin and cisplatin individually down-regulated the expression of nuclear factor κB (NF-κB). Effect of hesperidin or cisplatin on the protein expression levels of NF-κB expression in A549 and H460
The administration of hesperidin and cisplatin to A549 and H460 lung cancer cells resulted in notable decrease in the protein expression of NF-κB, with the lowest values observed in the hesperidin-treated H460 lung cancer cells. The experiment was performed in triplicates. The results presented are means of 2 independent experiments. aP < 0.01 vs. control bp < 0.01 vs. cisplatin-treated cells. NF-κB – nuclear factor κB
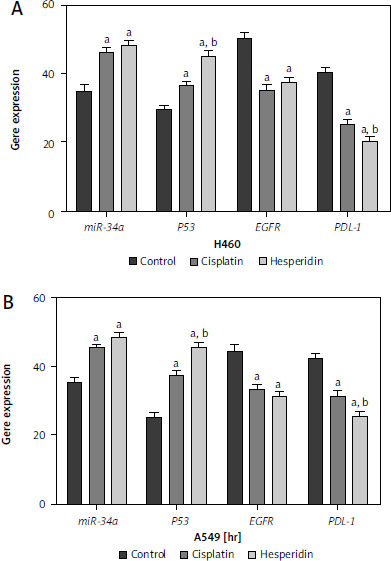
Hesperidin and cisplatin individually up-regulated MiR-34a and P53 while down-regulating epidermal growth factor receptor and programmed cell death ligand-1mRNA expressions in the neoplastic cells in A549 and H460 cells
To prove the mechanism by which hesperidin boosts the apoptosis process in A549 and H460 cells via targeting the miR-34a/PD-L1 signalling pathway, MRNA expressions of miR-34a, PD-L1, P53, and EGFR genes were assayed using reverse transcription PCR (RT-PCR). The polymerase chain reaction results of the negative regulator for invasion and metastasis; miR-34a in hesperidin-treated A549 revealed a significant expression of miR-34a in comparison with control cells, while cisplatin showed a lower expression of miR-34a in comparison with the hesperidin- treated H460 cells in addition to a significant boost in P53 expression in both hesperidin-treated A549 and H460 cells compared to cisplatin-treated and untreated cells, suggesting that hesperidin exerts a strong apoptotic effect via activation of tumour suppressor factors – P53 and miR-34a (Fig. 9). Additionally, the hesperidin-treated A549 presented a significant reduction of EGFR expression in A549 and a weaker expression of EGFR in H460 compared with H460 cells treated with cisplatin. Furthermore, the hesperidin- treated cells showed a significant down-regulation of the expression of the immune checkpoint inhibitor PDL-1 in both A549 and H460 cells in comparison with that in cisplatin-treated and untreated cells (Fig. 9). The findings mentioned above showed that hesperidin promoted apoptosis via up-regulation of P53 and miR-34a, thereby increasing the therapeutic value of hesperidin in the treatment of lung cancer cells.
Fig. 9
Hesperidin and cisplatin individually enhance the apoptosis process in H460 and A549
Polymerase chain reaction results of the negative regulator for invasion and metastasis; miR-34a in hesperidin-treated A549 revealed a significant expression of miR-34a compared to the control cells, while cisplatin showed a lower expression of miR-34a compared to the c-treated H460 cells. Representative chart showed a significant increase in P53 expression in both hesperidin-treated A549 and H460 cells compared to cisplatin-treated cells, suggesting that hesperidin exerts a strong apoptotic effect via activation of tumour suppressor factors; P53. Hesperidin-treated A549 showed a significant decrease of epidermal growth factor receptor (EGFR) expression in A549 and a weaker expression of EGFR in H460 compared with cells treated with cisplatin. Hesperidin-treated cells showed a significant down-regulation of the expression of the immune checkpoint inhibitor PDL-1 in both A549 and H460 cells compared with that in cisplatin-treated and control cells. The results presented are means of 2 independent experiments. The experiment was performed in triplicate. ap < 0.01 vs. control, bp < 0.01 vs. cisplatin-treated cells. EGFR – epidermal growth factor receptor, PDL-1 – programmed cell death ligand-1
Discussion
Today, cancer is the second most frequent reason for death globally. Cisplatin is the first-line therapy utilised for patients with lung cancer, despite recent advancements in new cancer treatment tactics [38]. Despite being effective against a variety of cancers, cisplatin has several limitations because of problems and unfavourable side effects. Resistance often occurs in cisplatin-treated patients, which poses a significant issue in the therapeutic environment [39]. The management of the cytotoxic side effects of chemotherapy medicines is essential to overcome these difficulties. Recently, it has been discovered that several natural compounds made from herbs and plant extracts have chemo-protective characteristics against the development of carcinogenesis [40].
Hesperidin is a member of the flavone family, which derives its members from citrus fruits. Bioflavonoids found in citrus seeds have been linked to apoptosis activation in human cancer cells earlier in in vitro experiments [41]. Recently, hesperidin has become a topic of discussion due to its effects on cancer, particularly its anticancer properties, which have been investigated in prior studies [42, 43]. Hesperidin regulates the production of proteins necessary for cell cycle progression that leads to mitochondrial apoptosis in lung cancer A549 cells, according to a prior study [44]. Hesperidin may be a potential cure for NSCLC based on these earlier findings [45]. The current investigation used cytotoxicity test, wound healing assay, and flow cytometer analysis to compare the antitumour effectiveness of hesperidin vs. cisplatin against NSCLC and to assess how both treatments affect cell death and proliferation.
Hesperidin’s cytotoxicity effects on lung cancer cells were evaluated in the current study using an SRB proliferation assay on A549 and H460 lung cancer cells. Our results demonstrated that hesperidin had no toxic effects on lung cells at a concentration of 100 µM in A549 and H460 cells; these effects were markedly amplified when the hesperidin concentration was raised to 1000 µM. In cells treated with cisplatin, the toxic effect began at 10 µM, demonstrating its impact on the feasibility of lung cancer cells. However, the powerful antitumour effectiveness of cisplatin is accompanied by multiple significant side effects and the development of chemo-resistance in cancer cells. Nephrotoxicity is the main side effect of cisplatin therapy, which develops because of the drug’s impairment of renal function and kidney tubular obstruction [46].
It is widely known that various chemotherapy medicines disrupt the cell cycle in a different manner [47]. Recent research revealed that the cell cycle selectively halted phase or phase transitions including G0/G1 or G2/M by a drug-induced apoptosis mechanism [48]. Cell cycle distribution may therefore be important for creating possible chemotherapeutic medicines. A study by Kabała-Dzik et al. showed that hesperidin at concentrations of 50–100 µM for 24–48 hours can increase cell population in the G0/G1 phase and the G2/M phase in MCF-7 cells. At a higher concentration of 100 µM, hesperidin caused induction of MCF-7 cells accumulation in the G0/G1 phase, which reflects a G0/G1 phase arrest [49]. Our results were consistent with those reported by another study [50], which demonstrated that hesperidin significantly decreased proliferation and began cell cycle arrest in lung cancer cells at the sub-G1 phase [51]. Hesperidin showed high peak concentration at the sub-G1 phase in A549 and G2 phase in H460 cells. Additionally, Aggarwal et al. [52] revealed that in addition to its anti-inflammatory and antioxidant properties, hesperidin’s principal mechanisms of action against cancer cells involve stoppage of the cell cycle and activation of apoptosis.
Additionally, Western blot and RT-qPCR analysis were applied to determine how hesperidin affected the miR-34a/PD-L1/EGFR signalling pathway. The co-inhibitory checkpoint signalling that regulates T-cell activation is greatly prompted by the engagement of PD-1, which is encoded by the PDCD1 gene and expressed on T-cells and PD-L1, which is encoded by the CD274 gene [53]. Numerous cancer forms produce high PD-L1 expression levels and use PD-L1/PD-1 signalling to disrupt T-cell immunity. Multiple studies found that inhibiting this route may be the gold standard treatment for the development of novel immune checkpoint blockade (ICB) because it has demonstrated antitumour effects in patients with advanced malignancies [54]. Regarding the ameliorative potency of hesperidin, our most recent research found that hesperidin blocked the NF-κB signalling pathway, which in turn decreased the expression of PDL-1 and elevated the expression of miR-34a in tumour cells, boosting the tumour suppressive effects on cancer cells. Hesperidin may be able to repair the negative feedback between miR-34a and inflammatory cytokines, as confirmed by the findings of other investigations [55]. Interestingly, another study demonstrated that miR-34a stimulates the signalling of the anti-inflammatory pathway, concurring with the study that showed that miR-34a is involved in regulating the production of proteins that reduce inflammation [56].
According to another study, hesperidin administration decreased PD-L1 expression in cancer cells, which enhanced immunosurveillance and decreased PDL-1 non-immune checkpoint activity, which attenuated the DNA damage response [57]. Our findings are in line with the study’s results of a prior study by Donia et al. [58], who hypothesised that hesperidin’s capability to halt tumour growth by inhibiting the expression of NF-κB may confer anticancer capabilities. However, recent research has shown that dysregulation of miRNA expression in many cancer types increases immune evasion and tumour spread [59]. Furthermore, lung cancer dysregulated miR-34a, which has a negative correlation with PD-L1 expression. Our investigation showed that p53 was inhibited when the PDL-1 signalling pathway was active in tumour cells, affecting the proliferation activity by losing the tumour suppressor ability of p53 [60], which is consistent with our findings.
Moreover, miRNA expression appears to be one of the processes causing cancer immune evasion, deregulation of p53 function in cancer cells is one such mechanism that is down-regulated, which is remarkable because p53 up-regulates miR-34a [61]. According to our findings, in both cancer cell lines p53 expression levels were considerably higher in cells treated with hesperidin in comparison with those treated with cisplatin. Furthermore, when compared to cisplatin therapy, hesperidin administration led to a much lower amount of PDL-1 expression, as demonstrated by our findings. Hesperidin treatment significantly reduced the accumulation of EGFR, whereas cisplatin treatment increased EGFR expression. This finding suggests that hesperidin gradually restored the inhibitory effect of miR-34a on inflammation and proliferation activity.
Prior studies suggest that the anticancer properties of cisplatin may be connected with the interaction of DNA molecules with cisplatin, which results in the production of free radicals that kill malignant cells [62]. However, the powerful antitumour effect of cisplatin is frequently accompanied by the emergence of tumour cell chemoresistance and several dose-limiting adverse effects [63]. In brief, the current findings regarding the ameliorative and cytoprotective potency of hesperidin showed that the administration of hesperidin alone can cause a significant decrease in tumour growth with cytoprotective properties in lung cancer cells, which was in agreement with another study that confirmed that these results are important signs of the powerful anticancer activities of hesperidin vs. cisplatin treatment in cancer cells [64]. However, some limitations of our research should be mentioned: it is also important to detect the miR 34a/PD-L1/NF-κB expression in other lung cancer different cell lines and to verify the applicability of the anti-lung cancer effects of hesperidin.
Conclusions
The current study showed that hesperidin administration may reduce lung cancer by encouraging apoptosis and reducing the growth of NSCLC cells via the miR-34a/PDL-1/NF-κB pathway. The strengths of this study lie in presenting hesperidin as a successful method and a secure cancer treatment that reduces the cytotoxicity impact linked to cisplatin and other chemotherapeutic medications. Hesperidin may present a new method for the treatment and prevention of lung cancer by avoiding some side effects of traditional cisplatin. However, the antitumour effect and mechanism of hesperidin require further study. With the research and development of hesperidin analogues and further study of their antitumour mechanisms, hesperidin may become a promising candidate compound for the prevention and treatment of tumours.
Hesperidin may be utilised to treat human lung cancer as an adjuvant or chemotherapy drug, even if its antitumour effects on lung cancer still need to be proven. The underlying molecular mechanisms’ in vivo pathways need more research. This research lies in the need to further apply the study results to the clinical settings in future studies.