
 ENGLISH
ENGLISH





Neurofibromatosis type 1 (NF1), also referred to as von Recklinghausen disease or von Recklinghausen syndrome, is a developmental disorder classified under phakomatoses. It is characterized by the presence of distinctive hamartomatous nodules and abnormalities in the organ of vision. Hamartomas are generally benign tumors, composed of tissues normally present in a specific organ, but occurring in excess and with an irregular distribution. The term ‘hamartoma’ is derived from the Greek word hamartia (‘error’), which highlights that hamartomas are the result of developmental error. Phakomatoses are a group of disorders that arise during the 3rd to 4th week of fetal life. The observed changes result from pathologies associated with the process of gastrulation during embryonic development, leading to abnormal transformation of the three embryonic layers – ectoderm, endoderm, and mesoderm – into body tissues and organs. Since developmental disorders affect various organs originating from the structures of these germ layers, NF1 is classified as a heterogeneous disease.
Other phakomatoses include Sturge-Weber syndrome (encephalotrigeminal angiomatosis), tuberous sclerosis (Bourneville disease, sclerosis tuberosa, epiloia), and capillary angiomatosis of the retina and cerebellum (von Hippel-Lindau disease).
The global prevalence of NF1 is estimated at 1 : 3,000 to 1 : 3,500 [1, 2]. The concept of phakomatoses was introduced to ophthalmology by the Dutch physician Jan van der Hoeve in 1932. He used it to describe a pathology in the organ of vision associated with the presence of hamartomatous lesions.
An important role in the development of hamartomas in NF1 is attributed to genetics [2]. Neurofibromatosis type 1 is an autosomal-dominant genetic disorder with complete penetrance. NF1 is not only a congenital condition; it also arises due to de novo mutations, with both types of abnormalities occurring in equal proportions. De novo mutations are found in chromosomes inherited from the father, and their number increases with the father’s age. The frequency of these mutations is higher in older fathers at the time of conception. Pathological changes in NF1 arise from mutations in a single gene located on chromosome 17q11.2. Neurofibromin, the protein encoded by this gene, belongs to the family of proteins activating the enzymatic hydrolysis of guanosine triphosphate (GTP) mediated by glyceraldehyde 3-phosphate (GAP), which stimulates the activity of guanosine triphosphate hydrolase, located in the RAS family on chromosome 6p21.2 [3]. RASopathies are developmental syndromes resulting from genetic alterations, germline mutations, and occasionally somatic mosaicism in genes that affect the RAS subfamily. Neurofibromin is found in various body tissues including the spleen, thymus, kidneys, and brain. Mutations in the NF1 gene cause a reduction or loss of protein function, disrupting cellular homeostasis and leading to uncontrolled activity of the RAS pathway. This results in unchecked cell proliferation and the clinical features of NF1 [4, 5].
Ophthalmic manifestations of NF1 are diverse, involving both extraocular and intraocular lesions, and appear at different stages of child development. Characteristic features are defined in international standards [6-8]. The clinical presentation of NF1 is a combination of developmental defects, systemic diseases, and neoplastic abnormalities (dysplasia and/or neoplasia) with lesions affecting the skin, bones, and blood vessels. A correct diagnosis of NF1 is determined by the presence of two or more of the clinical manifestations listed below, comprising ophthalmic symptoms. They include:
At least six café-au-lait macules (‘CALM’), > 5 mm in diameter in pubertal individuals and > 15 mm in postpubertal individuals. Café-au-lait macules are flat, skin-level spots, light to dark brown in color, resembling coffee with milk. They may have various shapes and sizes, and are hairless.
At least two neurofibromas of any type, or one plexiform neurofibroma. Neurofibromas are among the fundamental diagnostic clues for NF1, present in nearly all (approximately 99%) patients with the disorder. They are benign tumors that arise from Schwann cells which create myelin sheaths around peripheral nerve neurons, increasing the speed of nerve impulse transmission. Located in the skin, they usually develop in late childhood or early adolescence, rarely cause pain or neurological issues, and do not transform into malignant tumors. Neurofibromas can also occur in the brain, spinal cord, nerves, and various internal organs. Plexiform neurofibromas, unlike the previously mentioned localized neurofibromas, originate from the sheaths of multiple nerves and often extend beyond the nerve itself. Plexiform neurofibromas are primarily congenital and can be visible already at birth or emerge during early adolescence and young adulthood. Affecting approximately 30% of patients with NF1, they are primarily found in the facial region, as well as in the paravertebral area, mediastinum, and retroperitoneum. They are benign tumors; however, the rapid growth of plexiform neurofibromas and the associated pain can signal a potential transformation into malignant peripheral nerve sheath tumors. Located near the eyelids, combined with hemifacial atrophy, they deform these structures and may also cause amblyopia by obliterating the palpebral fissure. Plexiform neurofibromas may also cause dysplasia of orbital bones, potentially leading to compression of the optic nerve.
Freckling in the axillary or inguinal region, and occasionally on the back of the neck (in areas not exposed to sunlight), are another distinctive symptom of NF1. These pigmentation disorders typically manifest at approximately 2 to 3 years of age. They affect 81% of 6-year-old and 99% of 7-year-old children with NF1. The lesions are 1 to 3 mm in diameter, brown in color, and they typically occur in clusters [9].
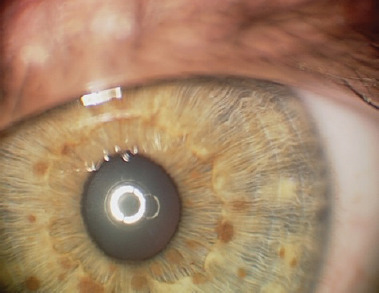
The presence of two or more Lisch nodules (iris hamartomas) is pathognomonic for NF1. Ophthalmologists assessing children or adults with confirmed or suspected NF1 are most frequently asked about the presence of Lisch nodules. The lesions present as small, raised, avascular nodules on the iris, measuring 0.1–2 mm in size, yellow-brown in color, with a gelatinous texture. They are visible on slit lamp examination (Figure 1).
They affect 10–90% of children with NF1, and their number rises with the child’s age. Lisch nodules generally do not affect visual acuity and are often asymptomatic [1]. They are not cancerous in nature, but may cause cosmetic concerns that negatively impact the patients’ quality of life [14]. Ultraviolet (UV) radiation from sunlight is believed to play a significant role in the development of Lisch nodules. The uneven distribution of Lisch nodules in the iris is hypothesized to be due to the relative protection of the upper half of the iris from UV radiation by the eyelid and eyebrow. The variation in the occurrence of Lisch nodules between light and dark irises is likely attributed to the protective role of pigmentation. Thus, genes responsible for iris color may be viewed as modifiers of the number and prominence of Lisch nodules in NF1 patients. The presence of Lisch nodules in the iris of one eye makes it highly probable that they also occur in the iris of the other eye. Multiple unilateral Lisch nodules occur rarely, and their presence may be explained by somatic mosaicism [15]. The appearance of Lisch nodules is primarily linked to the color of the iris. No significant correlation has been found between Lisch nodules and neurofibromatosis or the presence of CALM [16]. Histologically, Lisch nodules are consistent with melanocytic hamartomas of the iris [14].
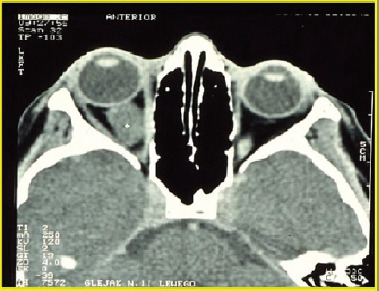
Optic glioma is a notable symptom of NF1 [10]. The tumor affects 15-20% of children under six years of age with neurofibromatosis type 1, with 70% of cases developing by the age of 10. It is more common in females, occurring in 25-50% of cases. Usually unilateral, a glioma can develop in any part of the visual pathway (Figure 2).
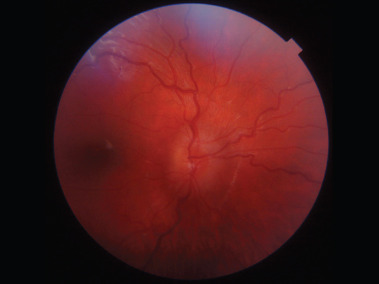
It relatively often develops in the optic chiasm area. Histologically, optic gliomas are most frequently identified as pilocytic astrocytomas (spongiomas) [2]. It is the most benign type of glioma in children and young adults, and it typically affects the anterior visual pathway. As a rule, it does not invade adjacent tissues and does not evolve into more malignant forms of glioma. Since young children do not always report changes associated with glioma, the tumor can remain silent and asymptomatic in its early stages. Slow increase of exophthalmos on the same body side as the developing glioma may also go unnoticed for a long period by those close to the child. Asymmetry in the eye sockets may only become apparent when observed by someone unfamiliar with the affected child. Just one-third of patients with glioma exhibit symptoms directly related to the tumor. The development of glioma results in impaired visual acuity, decreased vision in 35-50% of children, strabismus, exophthalmos, diplopia, and optic disc edema followed by its atrophy (Figure 3).
As glioma grows, it can compress the optic chiasm, which not only reduces visual acuity but, even more importantly, narrows the field of vision and potentially leads to irreversible vision loss [11-13]. These changes can significantly hinder a child’s social interactions and create challenges in performing activities [12]. If a glioma infiltrates the hypothalamus, it can lead to early onset of puberty. Patients with NF1 may also have other tumors of the visual system, such as neuromas and meningiomas.
Another manifestation of NF1 is distinctive bone damage, which can include sphenoid dysplasia or thinning of the bone cortex, sometimes accompanied by pseudoarthrosis. In the course of NF1, damage to the sphenoid bone can lead to distortion of the orbital area, resulting in pulsatile exophthalmos due to the absence or defect of the greater wing of the sphenoid bone, without orbital bruits. This damage can also cause cerebral herniation into the sphenoid sinus and orbit [9]. Patients with NF1 have also been diagnosed with retinal glial hamartomas and pigmented choroidal nevi, and have a greater prevalence of malignant choroidal melanoma.
Having a first-degree relative diagnosed with NF1 based on the specified criteria is a significant diagnostic indicator for this genetically inherited condition.
In recent years, choroidal anomalies (CA) have been recognized as a new ophthalmic manifestation of NF1 and incorporated into the diagnostic criteria. CA are oval-shaped lesions consisting of proliferative Schwann cells, melanocytes derived from the neural crest, and ganglion cells surrounding axons, all arranged in lamellar patterns. The prevalence and number of choroidal anomalies vary with age, being less frequent in children than in adults. CA occurs in 60.5% to 78.9% of children up to 12 years old, and in 86% to 100% of adults [9]. Choroidal anomalies cannot be detected using conventional ophthalmological examinations or fluorescein angiography. They are found in 100% of NF1 patients by infrared light examination with a scanning laser ophthalmoscope. The presence of CA as a clinical sign of NF1 is considered more accurate than the presence of Lisch nodules [17].
Glaucoma associated with NF1 can develop due to abnormalities in the embryonic development of the anterior chamber angle (congenital glaucoma) or from the closure of the filtration angle by phakomata in the angle itself, in the iris and the ciliary body. The condition is rare, typically ipsilateral, and often coexists with eyelid neurofibromas and facial lesions. Changes in the filtration angle may coexist with the presence of congenital ectropion uveae. In rare instances, neovascular glaucoma has also been noted. Mutations within the NF1 gene can trigger hyperproliferation of perivascular and endothelial cells, resulting in elevated vascular endothelial growth factor (VEGF) levels in response to retinal ischemia. This leads to reactive fibrovascular proliferation, glial cell and blood vessel damage, and secondary glaucoma [19].
Consequently, ocular changes are regarded as an important diagnostic feature of NF1.
Literature-reported lesions referred to as unidentified bright object (UBO), observed in brain magnetic resonance imaging (MRI) scans, do not impact visual sensation [7, 18].
The diagnosis of NF1 relies on clinical criteria established as early as 1988, which encompass a detailed medical history, a comprehensive ophthalmic examination, and additional tests, primarily genetic analysis and diagnostic imaging. Genetic diagnosis is not necessary in all cases [4]. Prenatal tests are mainly conducted in children whose parents have been diagnosed with NF1, as well as in children without a family history of the condition but presenting with café-au-lait macules. In the youngest patients, until the appearance of freckling in skin folds, the diagnosis of NF1 requires molecular testing. When diagnosing optic glioma, a combination of clinical evaluation and diagnostic imaging is essential, with computed tomography (CT) and especially MRI playing a prominent role.
There is no causal treatment for systemic abnormalities and ocular changes. Pediatric patients with NF1 require systematic medical monitoring by a broad team of collaborating specialists due to the heterogeneity of lesions and their varying onset times. It is also essential to provide psychological support to affected children. Management involves regular monitoring of disease progression and implementing therapies based on current indications [20, 21]. Pharmacological, oncological and surgical methods may be used, depending on the individual needs of the patient. The main goal of ophthalmological treatment is to maintain or improve visual acuity, making regular eye examinations crucial for patients. In children with glioma, individualized treatment is particularly important. Pediatric patients are primarily treated with chemotherapy, radiotherapy, or a combination of both. The first-line chemotherapeutic agents include vincristine and carboplatin, followed by vinblastine, vinorelbine, and temozolomide [5]. Surgical intervention for optic glioma is only recommended in specific situations, such as cases of a non-sighted eye with disfiguring exophthalmos, or with tumor growth that may infiltrate vital areas, such as conditions where the tumor threatens to damage the optic chiasm area. Despite significant advances in genetics, no genetic therapy for NF1 is currently available. However, such options are likely to emerge in the near future. With close follow-up and access to modern treatment modalities, children with NF1 are better able to participate in school activities and other important aspects of life.
In conclusion, NF1 is a genetically determined disorder regarded as the most prevalent phakomatosis. Neurofibromatosis type 1 presents with characteristic ocular symptoms developing both outside and/or inside the eye. Children with NF1 require continuous specialist medical care. There is no cure for this genetic disorder; treatment focuses solely on managing the symptoms. Genetic counseling and research in this field are crucial for diagnosing and managing both systemic and ocular lesions in NF1.