

Introduction
Asthma is a prevalent chronic inflammatory condition affecting the airways [1], with at least 300 million asthma patients worldwide, and the incidence is increasing yearly [2]. Asthma is associated with the pathology of various inflammatory mediators such as interleukin (IL)-4, IL-8, IL-17, and others [3]. In addition, asthma also involves multiple inflammatory cells such as T lymphocytes, eosinophils, neutrophils, and airway epithelial cells. Neutrophils are key players in the immune system and may respond rapidly to tissue damage and infections [4]. Neutrophils are recruited to allergic reaction sites initially and are significantly elevated in the airway secretions of severe asthma patients [5, 6]. One of the functions of neutrophils is the release of extracellular traps (NETs), which are web-like structures of DNA. The dysregulation of NETs may be associated with the pathogenesis of asthma [7]. Xia et al. concluded that neutrophil activation and NETosis are the main drivers of airway inflammation in a murine model of neutrophilic asthma [8]. Therefore, an in-depth exploration of the mechanisms of neutrophils in asthma may provide new insights into the treatment of asthma.
An increasing number of studies demonstrated a close relationship between autophagy and asthma [9, 10]. Autophagy is a self-degradation process that helps maintain cellular homeostasis and is significantly involved in the pathogenesis of asthma and in regulating the airway epithelial immune microenvironment [3, 11]. In asthma, autophagy’s regulation of immune cells is extensive [12]. Increased autophagy is involved in promoting type 2 immune response and eosinophilic asthma, while decreased autophagy may be important for the progression of neutrophilic asthma [13]. Therefore, understanding the potential molecular mechanisms that regulate neutrophil functions, such as autophagy [14], is crucial for targeted treatment and intervention strategies in asthma research.
In addition, the therapeutic application of src homology domain 2-containing protein tyrosine phosphatase 2 (SHP2) inhibitors in chronic respiratory diseases may also be promising. SHP2 belongs to the group of intracellular classical protein tyrosine phosphatases (PTPs) [15]. Researchers have indicated that SHP2 plays an important role in airway remodeling in asthma [15, 16]. Bone marrow SHP2-deficient mice have been shown to be unable to induce eosinophilia and airway hyper-responsiveness [17, 18]. However, the role of SHP2 in neutrophilic asthma remains unclear. The report by Ding et al. caught our attention. The report shows that psoriasiform skin inflammation is often accompanied by increased neutrophil infiltration. SHP2 promotes the formation of NETs in neutrophils through the ERK5 pathway, leading to the infiltration of proinflammatory cytokines and exacerbating psoriatic skin inflammation in mice [19]. Based on the background mentioned above, we hypothesized that SHP2 may aggravate asthma by promoting NET formation in neutrophils through the ERK5 pathway.
In our study, we collected peripheral blood from clinical asthma patients and built the dimethylsulfoxide (DMSO)-induced neutrophil-like cell model. Then, we investigated the role of SHP2 in neutrophils and its regulatory mechanisms. We expected the research to provide new insights into the complex regulatory effects of SHP2 on autophagy in neutrophils.
Material and methods
Clinical sample collection
We collected peripheral blood samples from healthy individuals and asthma patients. All asthma patients met the diagnostic criteria set by the Asthma Group of Chinese Thoracic Society [20], as follows:
Clinical symptoms and signs of typical asthma: 1) Recurrent wheezing, shortness of breath, with or without chest tightness or cough, frequent at night and in the morning, often related to exposure to allergens, cold air, physical or chemical stimulation, upper respiratory tract infection, exercise, etc. 2) Chronic persistent asthma during an attack and in some cases uncontrolled, with scattered or diffuse wheezing sounds in both lungs and a prolonged expiratory phase. 3) The above symptoms and signs can be relieved by treatment or spontaneously.
An objective examination of variable airflow limitation: 1) Positive bronchodilator test (FEV1 increase > 12% and absolute FEV1 increase > 200 ml after bronchodilator inhalation); or an increase in FEV1 of > 12% from baseline and an absolute increase in FEV1 of > 200 ml (excluding respiratory infections) after 4 weeks of anti- inflammatory treatment. 2) Positive bronchial provocation test: Inhaled challenges such as methacholine or histamine are typically used, and a 20% or greater decline in the FEV1 after the challenge is usually considered positive, indicating airway hyperresponsiveness. 3) The average diurnal variability of peak expiratory flow (PEF) (the sum of diurnal variability of PEF for at least 7 consecutive days/total days 7) > 10%; or PEF weekly variability {(highest within 2 weeks – lowest within 2 weeks) / [(highest within 2 weeks + lowest within 2 weeks) × 1/2] ×100%} > 20%.
Patients who met the above symptoms and signs and had any of the objective tests of airflow limitation, and for whom wheezing, shortness of breath, chest tightness and cough caused by other diseases were excluded, were diagnosed with asthma.
Inflammatory diseases in the lungs and other parts of the body, chronic obstructive pulmonary disease, tumors, blood system diseases, immune system diseases, coronary heart disease, hypertension, gastrointestinal ulcers with bleeding, intestinal perforation, epilepsy, and diabetes were excluded. The details are as follows:
Individuals with inflammatory diseases other than pulmonary conditions, such as dermatitis, myocarditis, myositis, and arthritis;
Patients with chronic obstructive pulmonary disease, including chronic bronchitis and emphysema;
Individuals with any type of tumor (benign or malignant);
Individuals with blood disorders, such as leukemia and lymphoma;
Patients with immune system disorders, such as autoimmune diseases, HIV infection, or AIDS;
Coronary artery disease or hypertension, including stable or unstable angina, myocardial infarction, congestive heart failure, myocardial infarction, early onset angina, pulmonary hypertension, and acute left heart failure;
Patients with gastrointestinal ulcers with active bleeding or severe complications such as intestinal perforation;
Patients with severe uncontrollable epilepsy or requiring specific drug treatments for epilepsy;
Individuals with poorly controlled diabetes or severe complications of diabetes.
Severe asthma patients required treatment with level 4 or 5 asthma medications (corticosteroids and long-acting β2-agonist) recommended by the Global Initiative for Asthma in the past year to maintain control or they still exhibited uncontrolled asthma despite the treatment above. Peripheral blood samples were collected using heparin vacuum tubes within 2 h of admission for subsequent experiments (3 tubes, 5 ml each). The use of human clinical samples for experimentation was approved by the Ethics Committee of The Fourth Hospital of Changsha (Approval Number: CSSDSYY-YXLL-SC-2023-01-05), and all participants provided informed consent. Based on the above requirements, the subjects could be divided into Healthy, Mild, and Severe asthma groups. Clinical sample information is provided in Table 1.
Table 1
Clinical sample information
Isolation and identification of neutrophils
Following the guidelines from the manufacturer, the Human Peripheral Blood Neutrophil Isolate Solution Kit (P9402, Solarbio, Beijing, China) was first used to isolate neutrophils in peripheral blood from clinical samples. Each group of cells was collected and centrifuged at 900 rpm for 5 min. Then, the cell pellet was resuspended in 100 µl of basal culture medium. An appropriate amount of CD11b (11-0112-82, eBioscience, San Diego, CA, USA) antibody was added. After incubation, the cells were centrifuged, and the supernatant was discarded. Finally, the assay was performed by a flow cytometer (A00-1-1102, Beckman, Brea, CA, USA).
HL-60 cell differentiation model
The human promyelocytic leukemia cell line HL-60 was derived from Abiowell (AW-CCH071, Changsha, China) and cultured in IMDM medium (iCell-0008, iCell Bioscience Inc, Shanghai, China) containing 20% FBS (10099141, Gibco, Grand Island, NY, USA) and 1% penicillin-streptomycin (SV30010, Beyotime, Shanghai, China) under conditions maintained at 37°C, 5% CO2, saturated humidity. HL-60 cells with a density of 106 cells/ml were differentiated into neutrophil-like cells using 1.3% dimethylsulfoxide (DMSO, D8371, Solarbio) for 6 days. Giemsa staining and RT-qPCR experiments were used to identify the status of cell transformation. Neutrophil-like cells were treated with 1 µM rapamycin (RAP, IR0010, Solarbio) for 48 h to activate autophagy [21].
To investigate the mechanism of SHP2 affecting the autophagy of neutrophil-like cells, the cells were divi- ded into four groups: 1) Control: Neutrophil-like cells were cultured normally. 2) RAP: Neutrophil-like cells were treated with 1 µM Rap for 48 h. 3) RAP + oe-NC: Neutrophil-like cells were treated with 1 µM Rap for 48 h and transfected with overexpression-negative control (oe-NC). 4) RAP + oe-SHP2: Neutrophil-like cells were treated with 1 µM Rap for 48 h and transfected with oe-SHP2.
To explore the effect of SHP2 on NET formation through the ERK5 pathway, neutrophil-like cells were divided into five groups: 1) Control: Neutrophil-like cells were cultured normally. 2) si-NC: Neutrophil-like cells were transfected with small interfering RNA-negative control (si-NC). 3) si-SHP2: Neutrophil-like cells were transfected with si-SHP2. 4) si-SHP2 + oe-NC: Neutrophil-like cells were transfected with si-SHP2 and oe-NC. 5) si-SHP2 + oe-ERK5: Neutrophil-like cells were transfected with si-SHP2 and oe-ERK5.
To investigate the effect of SHP2 on autophagy and NET formation in peripheral blood-derived neutrophils from asthma patients, the cells were divided into three groups: 1) Neutrophils: Neutrophils were cultured normally. 2) si-NC: Neutrophils were transfected with si-NC. 3) si-SHP2: Neutrophils were transfected with si-SHP2.
Cell transfection
When the degree of cell fusion reached 70-80%, cell transfection was carried out for 48 h using Lipofectamine 2000 (11668019, Invitrogen, Carlsbad, CA, USA). Neutrophil-like cells or neutrophils were transfected with oe-SHP2 (HG-HO002834), si-SHP2 (HG-HS 002834), oe-ERK5 (HG-HO002749) or their negative controls. All plasmid sequences were synthesized by HonorGene.
Enzyme-linked immunosorbent assay (ELISA)
The levels of IL-4, IL-8, and IL-17 in clinical blood samples and neutrophil-like cells were assessed according to the ELISA kit instructions. The Human IL-4 Kit (KE00016), IL-8 Kit (KE00006) and IL-17 Kit (KE00203) were all ordered from Proteintech. In addition, the dsDNA level in the cells was measured using the kit (YJ060072, YuanjuBio, Shanghai, China). Briefly, the steps were as follows: 100 µl of the enzyme-labeled reagent was added, followed by incubation for 60 min. After washing, 50 µl of chromogens were added. Finally, the absorbance values of each well were measured at a wavelength of 450 nm after the termination of the reaction.
Myeloperoxidase (MPO) activity in the cells was analyzed according to the manufacturer’s instructions (A044-1-1, njjcbio, Nanjing, China). Finally, the absorbance value was measured at a wavelength of 460 nm. MPO activity (U/l) = (measured value – control value) / 11.3*V.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells according to the Total RNA Extraction (Trizol) (15596026, Thermo, Waltham, MA, USA) method specification. Then, the absorbance values of total RNA at 260 nm and 280 nm were measured, and their concentration and purity were calculated. The samples were subjected to RT-qPCR in a real-time PCR instrument (QuantStudio 1, ABI, Foster, CA, USA) with SYBR Mix (15 µl), positive primer (1 µl), negative primer (1 µl), cDNA (2 µl), RNase Free ddH2O (11 µl). Reverse transcription products were obtained and detected in fluorescence. The 2-ΔΔCt method was used to calculate the relative expression of each gene of interest, and 3 complex wells were set up for each sample. GAPDH was used as the internal reference gene. Primer sequences are shown in Table 2.
Table 2
Primer sequences used in the study
Immunofluorescence (IF) staining
Suspended cells were spread evenly on the slide, and the slide was dried and rinsed with water. Then 0.3% Triton X-100 was used to permeate for 30 min. After washing, 5% BSA was added to the block for 60 min. Subsequently, appropriate diluted LC3 (1 : 50, 14600-1-AP, PTG, USA) was added to the slides and incubated overnight at 4°C. Then 50-100 µl of Goat anti-Rabbit IgG (H+L) Secondary Antibody Alexa Fluor488 (1 : 200, AWS0005c, Abiowell) was added and incubated with LC3 for 90 min.
In addition, an HRP secondary antibody was added after incubation with MPO (1 : 50, 22225-1-AP, PTG). TSA-570 (AWI0702b, Abiowell) was then added to the reaction mixture for 10 min. After the last round of antibody elution, an endogenous peroxidase-blocking agent was incubated in the dark for 10 min. After blocking with BSA, citrullinated histone 3 (cit-H3, 1 : 50, ab281584, Abcam, UK) was incubated. After the HRP secondary antibody and TSA-570 were added, the previous round of antibodies was eluted. DAPI working solution (AWI0429, Abiowell) was applied to stain the nucleus for 10 min, and the slices were sealed with glycerol. Finally, images were acquired by fluorescence microscopy (400×) and photographed. LC3 or MPO showed green fluorescence, cit-H3 showed red fluorescence, and DAPI showed blue fluorescence.
Western blot (WB) analysis
Cells from each group were collected and rinsed with pre-chilled PBS buffer, and RIPA lysate (AWB0136, Abiowell) was added to lyse the cells. After the protein concentration had been measured with the BCA kit (AWB0104, Abiowell), the corresponding volume of protein was added to the 5 × loading buffer (AWB0055, Abiowell) to mix. The electrophoresis constant voltage was 75 V, and the time was 130 min. The transfer current was 300 mA. After transfer, the membrane was rinsed once in 1×PBST, and then the membrane was completely immersed in the blocking solution and shaken for 90 min. Primary antibodies were diluted in a ratio with 1×PBST: beclin1 (BECN1, AWA00914, 1 : 1000, Abiowell), LC3II/I (AWA01019, 1 : 1000, Abiowell), p62 (66184-1-Ig, 1 : 10000, Proteintech), SHP2 (ab32083, 1 : 1000, Abcam), MPO (22225-1-AP, 1 : 1000, Proteintech), neutrophil elastase (ELANE, AWA42997, 1 : 1000, Abiowell), peptidyl arginine deiminase 4 (PADI4, 17373-1-AP, 1 : 1000, Proteintech), ERK5 (AWA00874, 1 : 1000, Abiowell), and β-actin (AWA80002, 1 : 5000, Abiowell). The membrane was incubated with the primary antibody at 4°C overnight and left at room temperature for 30 min the next day. After washing, HRP goat anti-mouse IgG (SA00001-1, 1:5000, Proteintech) and HRP goat anti-rabbit IgG (SA00001-2, 1 : 6000, Proteintech) were incubated with the membrane for 90 min. Finally, ECL chemiluminescence solution (AWB0005, Abiowell) was incubated with the membrane for 1 min and observed with a chemiluminescence imaging system (ChemiScope 6100, Clinx Science Instruments, Shanghai, China).
Giemsa staining
Cells from each group were collected and fixed by adding 1 ml of 4% paraformaldehyde solution for 30 min, and then 50 µl of Giemsa staining solution was used to the stain for 10 min. After washing for 30 s, they were observed under the microscope (DSZ2000X, Cnmicro, Beijing, China).
Cell Counting Kit-8 (CCK-8) assay
The CCK-8 kit (NU679, DOJINDO, Japan) was used to detect the viability of each group of cells. Cells from the logarithmic growth phase were seeded in 96-well plates with a density of 5 × 103 cells/well, 100 µl per well. After the corresponding culture time had elapsed, the medium was replaced, and 100 µl/well of CCK-8-containing medium was added. Finally, the optical density value at 450 nm was analyzed with a microplate reader (MB-530, Heales, Shenzhen, China), and the mean value was taken for a histogram.
Statistical analysis
All experimental data were analyzed with GraphPad Prism8.0. Each value was presented as the mean ± standard deviation. A parametric t test or one-way analysis of variance (ANOVA) was used to detect the significance of differences. The Pearson correlation test was utilized to analyze the correlation between variables. P < 0.05 was considered statistically significant.
Results
Decreased neutrophil autophagy in the peripheral blood of asthma patients
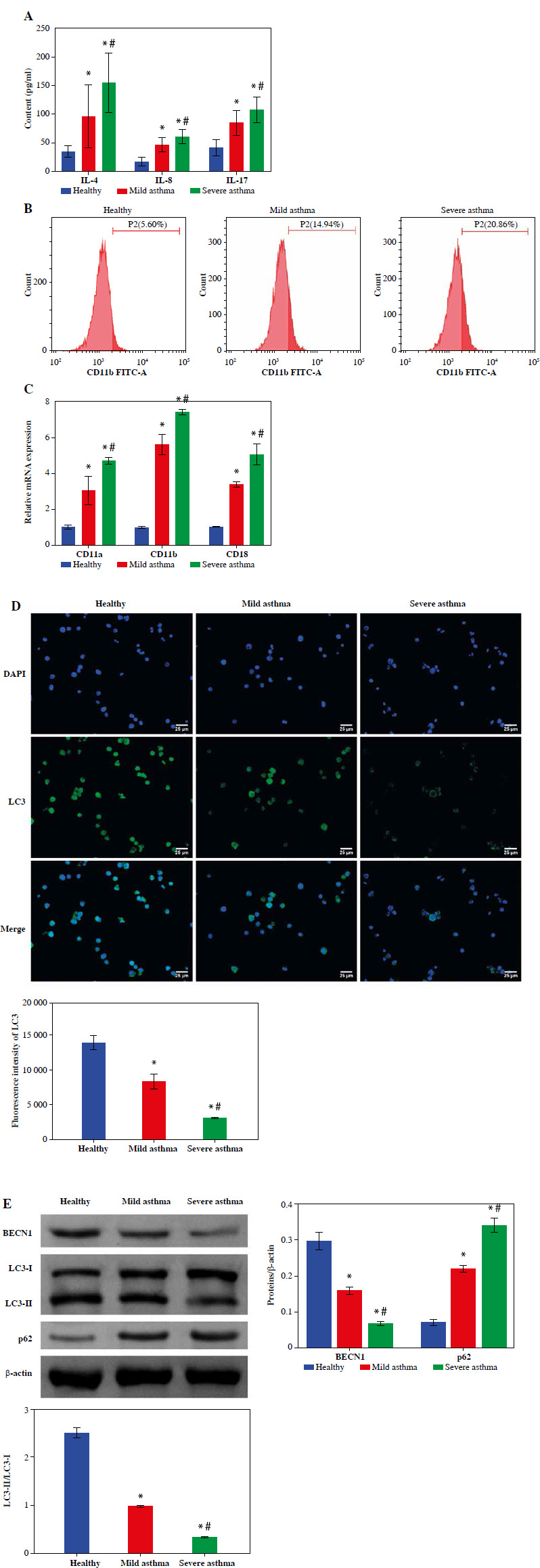
Blood biochemical test results showed that the percentage of neutrophils showed a gradual decrease when comparing the Healthy, Mild asthma, and Severe asthma groups. Because asthma is an inflammatory disease, we measured the levels of the inflammatory factors IL-4, IL-8, and IL-17 in the peripheral blood of asthma patients. ELISA results showed that IL-4, IL-8 and IL-17 levels were significantly higher in the Mild and Severe asthma groups compared with the Healthy group. Moreover, in the Severe asthma group it was significantly higher than the Mild asthma group (Fig. 1A). The percentage of neutrophils in the blood could be used to assess the severity of asthma [22]. Flow cytometry results indicated that the positive rate of CD11b was increased gradually with the severity of asthma (Fig. 1B). To validate the activation status of neutrophils in peripheral blood, RT-qPCR was conducted to assess the expression levels of the activation markers CD11a, CD11b, and CD18 on neutrophils. The mRNA expression of these markers was notably lower in the Healthy group than in the Mild and Severe asthma groups and significantly higher in the Severe asthma group than in the Mild asthma group (Fig. 1C). Subsequently, we evaluated the expression of the autophagy marker LC3 in neutrophils by IF. The expression of LC3 in the Mild and Severe asthma groups was significantly lower than that in the Healthy group. LC3 expression was lower in the severe group compared with the Mild group (Fig. 1D). It has been reported that both BECN1 and LC3 are classical autophagy-related proteins [23, 24], and the conversion from LC3-I to LC3-II has been commonly used to detect autophagy status [25]. Therefore, we detected the expression levels of BECN1, LC3 and p62 by WB. The results showed that the BECN1 expression and the LC3-I conversion level gradually decreased with the severity of asthma, while the p62 expression gradually increased with the severity of asthma (Fig. 1E). The above data indicated an abnormal increase in activated neutrophils, and autophagy was significantly reduced in patients with asthma.
Fig. 1
Decreased neutrophil autophagy in the peripheral blood of asthma patients. A) The contents of IL-4, IL-8 and IL-17 in serum were measured using ELISA. B) Flow cytometry was utilized to measure the percentage of neutrophils in peripheral blood. C) Expression of neutrophil activity markers CD11a, CD11b and CD18 was detected using RT-qPCR. *p < 0.05 vs. Healthy and #p < 0.05 vs. Mild asthma D) Expression of autophagy marker LC3 in neutrophils was detected by IF. (E) WB was used to analyze the protein expression of BECN1, LC3 and p62 in neutrophils. *p < 0.05 vs. Healthy and #p < 0.05 vs. Mild asthma E) WB was used to analyze the protein expression of BECN1, LC3 and p62 in neutrophils. *p < 0.05 vs. Healthy and #p < 0.05 vs. Mild asthma
NET formation of neutrophils was upregulated in the peripheral blood of asthma patients
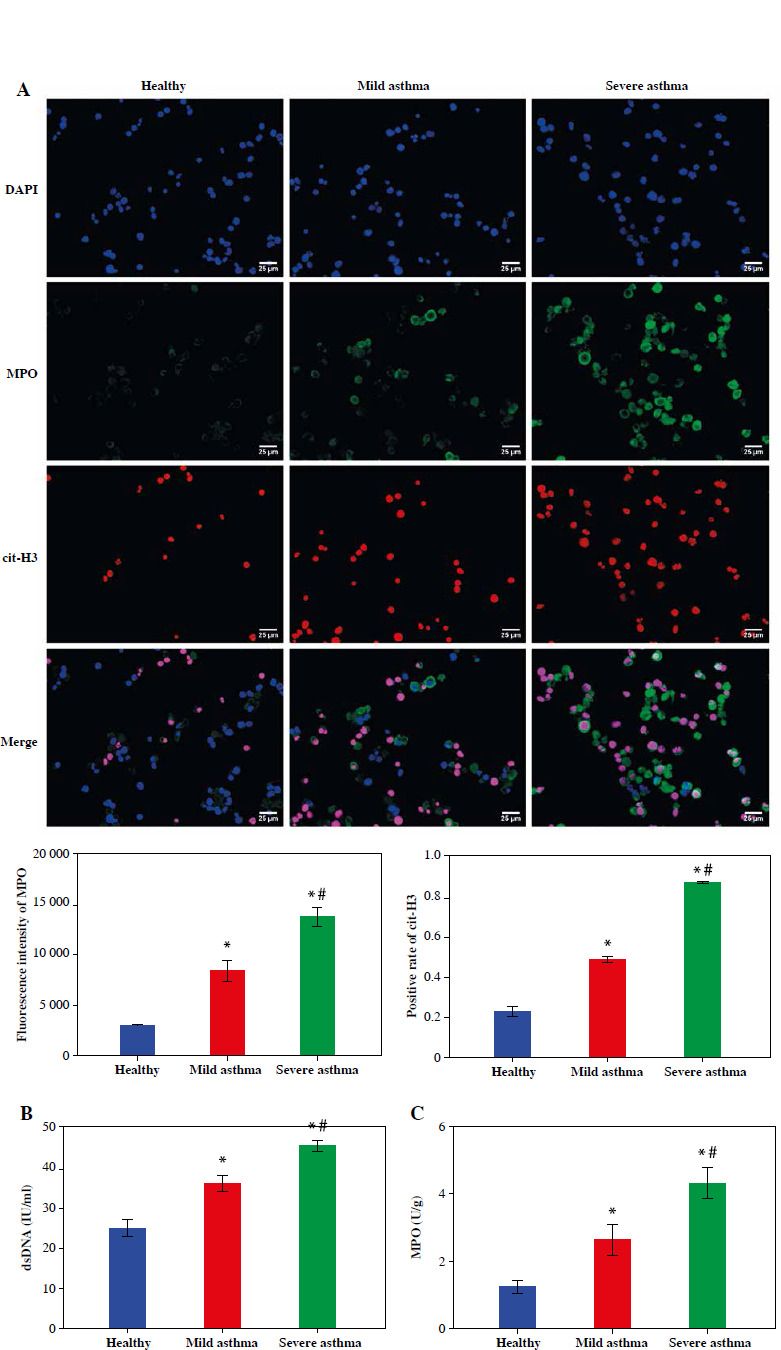
To further explore the formation of neutrophil NETs in asthma patients, we evaluated two key proteins for NET formation by IF. The results showed that the expression levels of MPO and cit-H3 were notably elevated in asthma patients and higher in the Severe asthma group than in the Mild asthma group (Fig. 2A). In addition, the dsDNA level was used to quantify NET production in the neutrophil supernatant of healthy individuals and patients with asthma. The results indicated that dsDNA levels in asthma patients were significantly higher than in the healthy individuals and increased significantly with asthma severity (Fig. 2B). We also assessed MPO activity in neutrophils. In the Mild asthma group, the MPO activity was significantly higher than that in the Healthy group, while in the Severe asthma group, the MPO activity was significantly higher compared to the Mild asthma group (Fig. 2C). Taken together, these findings showed that NET formation and neutrophils were closely associated with asthma development.
Fig. 2
NET formation of neutrophils was upregulated in the peripheral blood of asthma patients. A) IF was used to evaluate the expression of cit-H3 and MPO. B, C) Levels of dsDNA and MPO activity in neutrophils were detected by ELISA. *p < 0.05 vs. Healthy and #p < 0.05 vs. Mild asthma B, C) Levels of dsDNA and MPO activity in neutrophils were detected by ELISA. *p < 0.05 vs. Healthy and #p < 0.05 vs. Mild asthma
SHP2 was highly expressed in the neutrophils of asthma patients
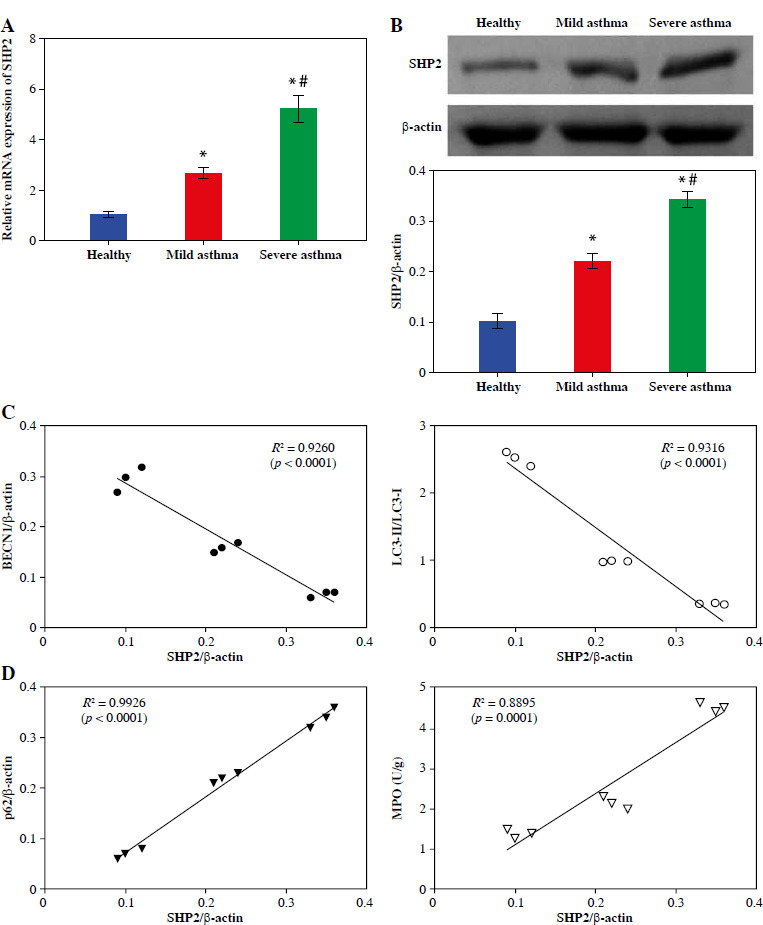
Subsequently, we assessed SHP2 expression in neutrophils from clinical samples. The RT-qPCR and Western blotting results indicated a gradual increase in both the mRNA and protein expression levels of SHP2 from the Healthy to the Mild and to the Severe asthma groups, as depicted in Figure 3A, B. Subsequently, to elucidate the association between expression of SHP2 and autophagy-related factors, Pearson’s correlation analysis was conducted to examine their correlations. As illustrated in Figure 3C, D, SHP2 exhibited a negative correlation with BECN1 and LC3 while showing a positive correlation with p62 and MPO.
Fig. 3
SHP2 was highly expressed in the neutrophils of asthma patients. A, B) RT-qPCR and WB were conducted to analyze the SHP2 expression. C, D) Pearson’s correlation test was used to analyze the correlation between SHP2 and BECN1, LC3, p62, and MPO. *p < 0.05 vs. Healthy and #p < 0.05 vs. Mild asthma
SHP2 inhibited autophagy in neutrophil-like cells
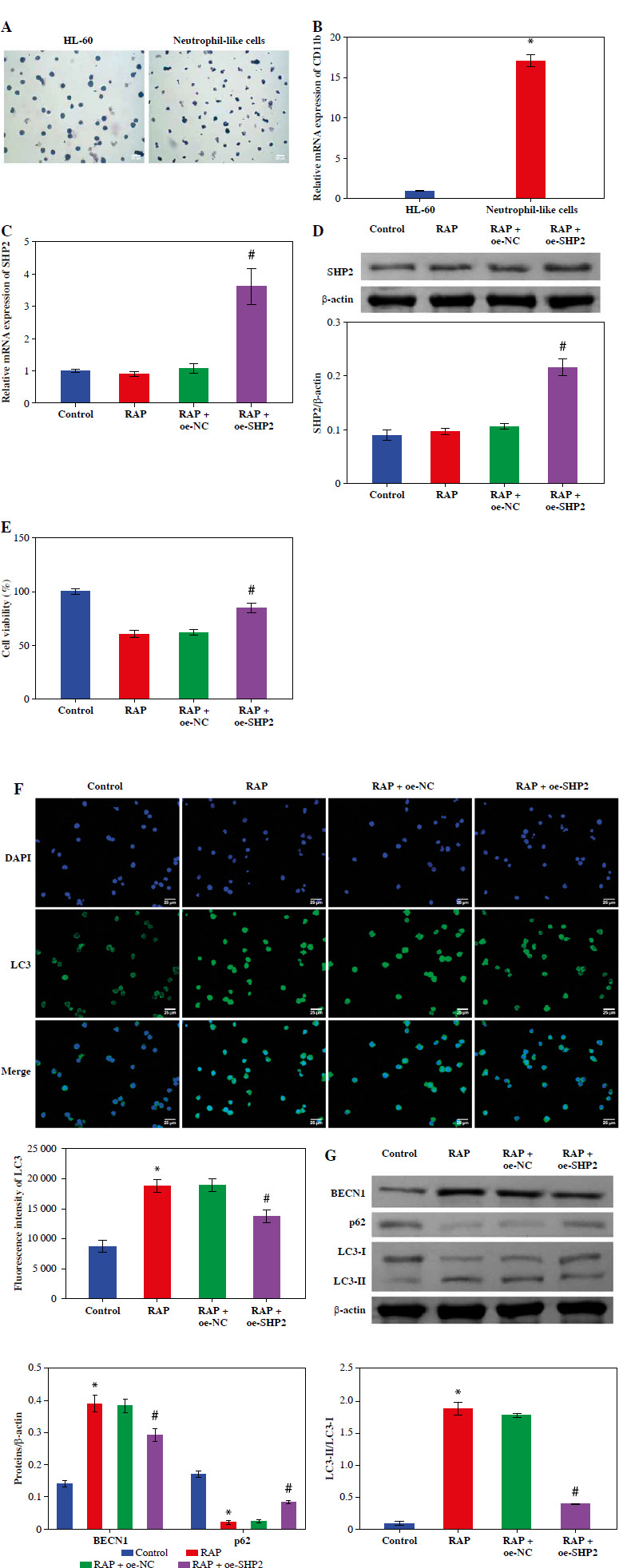
HL-60 and neutrophil-like cells were subjected to Giemsa staining, and microscopic examination revealed the distinctive polarized morphology of neutrophil-like cells, characterized by contracted tails and front wrinkles, as illustrated in Figure 4A. Additionally, CD11b was identified as a differentiation and maturation marker for neutrophils derived from HL-60 cells [26]. We examined the CD11b expression in cells after 6 days of DMSO treatment using RT-qPCR. In the Neutrophil-like cell group, the CD11b expression was significantly higher than in the HL-60 group (Fig. 4B). These data indicated successful induction of neutrophil-like cells. Then, we treated the cells with the autophagy inducer RAP. The RT-qPCR and WB results indicated that the expression level of SHP2 did not differ significantly from the Control group after RAP treatment. SHP2 expression was significantly higher in the RAP + oe-SHP2 group compared with the RAP + oe-NC group (Fig. 4C, D). This result verified the successful transfection of oe-SHP2. Subsequently, the CCK-8 assay showed that the viability of neutrophil-like cells in the RAP group was significantly lower compared with the Control group. After overexpression of SHP2, the cell viability was significantly increased (Fig. 4E). To further investigate the mechanism of SHP2 affecting the autophagy of neutrophil-like cells, we evaluated the autophagy of neutrophil-like cells through IF and WB. The results demonstrated that the expression levels of BECN1 and LC3 were increased after RAP treatment. Then, the expression levels of BECN1 and LC3 were significantly increased in the RAP + oe-SHP2 group. Correspondingly, the expression level of p62 in each group was opposite to the trend of BECN1 and LC3 (Fig. 4F, G). The above data indicated that SHP2 could show the autophagy of neutrophil-like cells.
Fig. 4
SHP2 inhibited autophagy in neutrophil-like cells. A) Cellular polarization morphology changes were observed through Giemsa staining. B) CD11b expression in cells after 6 days of DMSO treatment was detected by RT-qPCR. *p < 0.05 vs. HL-60. C, D) SHP2 expression level was analyzed in neutrophil-like cells by RT-qPCR and WB. E) CCK-8 assay was utilized to detect cell viability. *p < 0.05 vs. Control and #p < 0.05 vs. RAP + oe-NC F) Distribution of LC3 in neutrophil-like cells was detected using IF. G) Protein expression levels of BECN1, LC3 and p62 were detected in neutrophil-like cells using WB. *p < 0.05 vs. Control and #p < 0.05 vs. RAP + oe-NC
Inhibition of NET formation in neutrophil-like cells through down-regulating the SHP2/ERK5 pathway
To explore the effect of SHP2 on NET formation, we interfered with SHP2 expression in neutrophil-like cells. The RT-qPCR and WB results verified the success of si-SHP2 transfection (Fig. 5A). Subsequently, we examined some relevant indicators of NET formation. The expression levels of cit-H3, dsDNA level and MPO activity in the si-SHP2 group were lower than those in the si-NC group (Fig. 5B, D). In addition, we used WB to examine genes for proteins involved in the formation of NETs. The results indicated that the expression levels of MPO, ELANE and PADI4 were significantly decreased after interfering with SHP2 expression (Fig. 5E). At the same time, ERK5 protein expression was significantly lower in the si-SHP2 group than in the si-NC group (Fig. 5F). To further investigate whether SHP2 inhibits NET formation by suppressing ERK5 expression, we cotransfected si-SHP2 and oe-ERK5 and their negative controls in neutrophil-like cells. The RT-qPCR and WB results showed a significant increase in ERK5 expression but no significant change in the SHP2 after overexpression of ERK5 (Fig. 5G). This result indicated that SHP2 could regulate ERK5 expression. Subsequently, our findings revealed that ERK5 overexpression partially counteracted the inhibitory effect of si-SHP2 on NET formation in neutrophil-like cells (Fig. 5H-K). In summary, the above results indicated that SHP2 regulated NET formation through the ERK5 pathway in neutrophil-like cells.
Fig. 5
Inhibition of NET formation in neutrophil-like cells through down-regulating the SHP2/ERK5 pathway. A) RT-qPCR and WB were performed to detect SHP2 expression. *p < 0.05 vs. si-NC and #p < 0.05 vs. si-SHP2 + oe-NC B) IF staining was employed to detect cit-H3 expression. C, D) Levels of dsDNA and MPO activity in neutrophil-like cells were measured. *p < 0.05 vs. si-NC and #p < 0.05 vs. si-SHP2 + oe-NC E, F) Protein expression of MPO, ELANE, PADI4 and ERK5 was analyzed using WB. G) Expression levels of SHP2 and ERK5 were analyzed using RT-qPCR and WB. *p < 0.05 vs. si-NC and #p < 0.05 vs. si-SHP2 + oe-NC H) Cit-H3 expression was detected using IF. I, J) Levels of dsDNA and MPO activity in neutrophils were measured. K) WB was performed to analyze the protein expression of MPO, ELANE, and PADI4 in neutrophil-like cells. *p < 0.05 vs. si-NC and #p < 0.05 vs. si-SHP2 + oe-NC
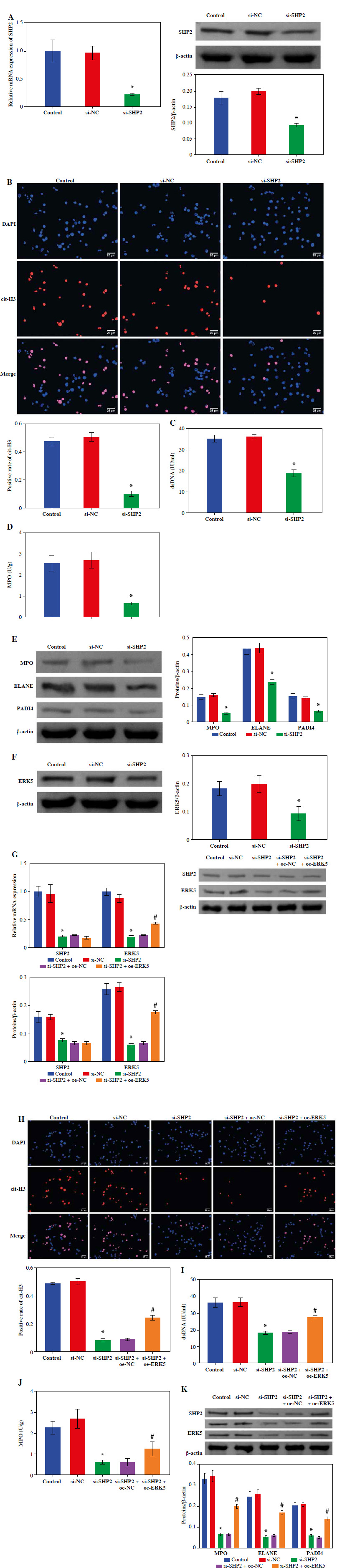
Down-regulation of SHP2 promoted autophagy and inhibited the formation of NETs in peripheral blood-derived neutrophils from asthma patients
We detected the expression of SHP2 and ERK5 in peripheral blood-derived neutrophils from asthma patients. As shown in Figure 6A, the expression levels of SHP2 and ERK5 were significantly lower in the si-SHP2 group compared with the si-NC group. After silencing the expression of si-SHP2, the distribution of LC3 and the protein expression levels of BECN1 and LC3 in the neutrophils were significantly decreased. The p62 protein expression in the si-SHP2 group was significantly lower compared to the si-NC group (Fig. 6B, C). The above results suggested that interfering with SHP2 expression inhibited autophagy in neutrophils from asthma patients.
Fig. 6
Down-regulation of SHP2 promoted autophagy and inhibited formation of NETs in peripheral blood-derived neutrophils from asthma patients. A) Expression levels of SHP2 and ERK5 were detected by RT-qPCR and WB in neutrophils. B) IF and WB were utilized to detect the expression of LC3, BECN1 and p62 to assess neutrophil autophagy. *p < 0.05 vs. si-NC C) IF and WB were utilized to detect the expression of LC3, BECN1 and p62 to assess neutrophil autophagy. D) Expression of cit-H3 was detected using IF. *p < 0.05 vs. si-NC E, F) Levels of dsDNA and MPO activity in neutrophils were measured by ELISA. G) WB was performed to analyze the protein expression of MPO, ELANE, and PADI4 in neutrophils. *p < 0.05 vs. si-NC
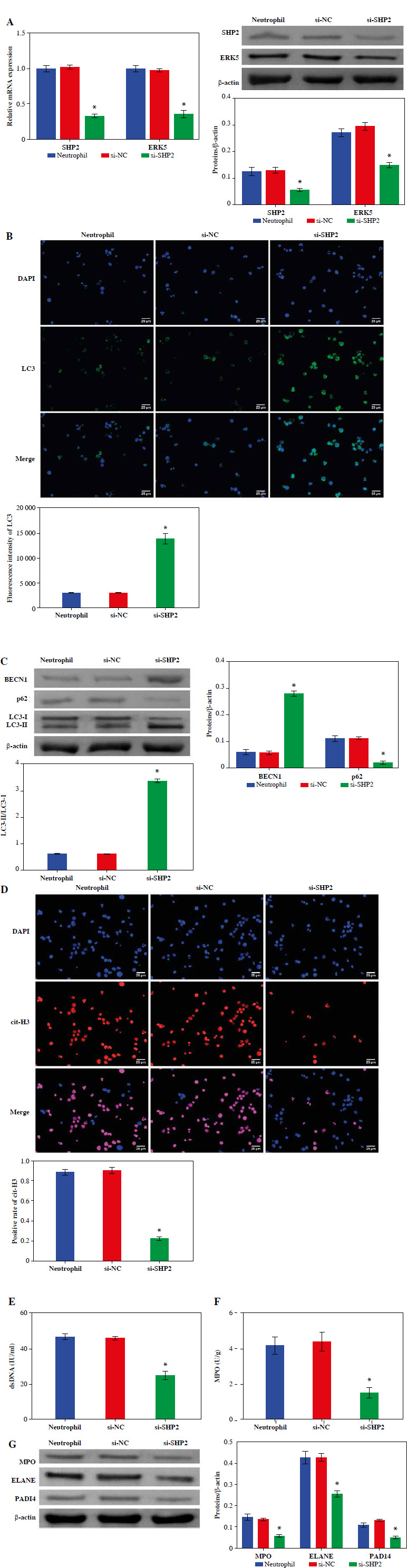
Moreover, we also evaluated NET formation using IF, ELISA and WB in neutrophils from asthma patients. The results demonstrated that cit-H3 expression, dsDNA level and MPO activity were significantly decreased after interfering with si-SHP2 (Fig. 6D-F). The protein expression levels of MPO, ELANE and PADI4 in the si-SHP2 group were significantly lower than those in the si-NC group (Fig. 6G). To sum up, our study demonstrated that SHP2 played an important role in regulating neutrophil autophagy and NET formation in asthma patients.
Discussion
Neutrophils are immune cells, and inappropriate or abnormal neutrophil activation harmed the host and led to autoimmune and inflammatory diseases [4]. Neutrophils in the airways of asthmatic patients were associated with disease severity and acute asthma exacerbations [27]. In this study, the levels of inflammatory factors IL-4, IL-8, and IL-17 were highest in the peripheral blood of patients with severe asthma, while the number of neutrophils gradually increased in blood samples from healthy to mild and severe asthma patients. These observations further validated that measuring peripheral blood neutrophils could serve as a method for assessing the severity of human asthma.
In our study, we examined the neutrophil autophagy in the peripheral blood of asthma patients. The results indicated that the autophagy-related factors BECN1, LC3 and p62 were abnormally expressed in asthma patients. These data suggested that autophagy of neutrophils was strongly associated with asthma, which was consistent with the findings of Quoc et al. [28]. In addition, autophagy activation correlated with NET production in neutrophils [29]. NET formation could occur through a cell death process known as NETosis or in association with the release of DNA from surviving neutrophils [30]. The study by Keir et al. demonstrated that NET was a key marker of disease severity and response to therapy in bronchiectasis [31]. Neutrophil autophagy and NET aggravated the severity of asthma by damaging the airway epithelium and triggering an inflammatory response [32]. Autophagy drove NET formation in neutrophils, thereby promoting pulmonary fibrosis [33]. In contrast, the autophagy activator mTOR reduced signs of pulmonary inflammation in the lungs of mice with cystic fibrosis [34]. Racanelli et al. suggested that autophagy might exert a more significant protective influence on airway inflammation and hyperresponsiveness within the framework of innate immune cell signaling [9]. Our data indicated that NET formation was markedly increased in neutrophils from asthmatic patients and that SHP2 was significantly correlated with autophagy and NET-related factors.
Subsequently, we found that SHP2 overexpression partially counteracted the inhibitory effect of RAP on cell viability. At the same time, our data also indicated the inhibitory effect of SHP2 on autophagy in neutrophil-like cells. Studies showed that SHP2 could be closely related to the autophagy mechanism. Inhibition of SHP2-related pathways by knocking out SHP2 or pharmacological inhibitors could significantly alleviate the inhibition of apoptosis and autophagy in mouse models of ovarian cancer [35]. Moreover, SHP2 could enhance autophagy in non-small cell lung cancer cells by promoting the MEK/ERK pathway, thereby promoting invasion and metastasis of non-small cell lung cancer cells [36]. Karampitsakos et al. demonstrated that SHP2 reduced the fibrotic response in a lung fibroblast cell line by inducing autophagy [37]. Findings have indicated that SHP2 may have different effects on autophagy in different diseases. Recently, it was reported that the suppression of SHP2 in human neutrophil lines enhanced the release of proinflammatory cytokines and NETs, underscoring the pathogenic impact of plasma- derived exosomal SHP2 deficiency in fueling neutrophil activation [38]. This was consistent with the results of our study. We interfered with SHP2 expression in neutrophil-like and neutrophil-like cells. We further verified that SHP2 could regulate autophagy and NET formation in neutrophils through ERK5.
ERK5, a member of the MAPK family, has been found to be involved in a variety of cellular functions and to be associated with NET formation [39, 40]. El-Deeb et al. suggested ERK5 as a molecular signal transducer involved in the regulation of autophagy in Parkinson’s rats [41]. The molecular mechanism of ERK5 in asthma has not yet been elucidated. Our study revealed that ERK5 overexpression partially counteracted the inhibitory effect of si-SHP2 on NET formation in neutrophil-like cells.
However, several limitations need to be addressed in future work. Firstly, the results of this study were obtained based on clinical blood samples and cell models, and we could not determine whether the SHP2/ERK5 pathway could directly act on asthma disease. Due to limited conditions, we were unable to collect more clinical samples. Therefore, it is imperative for future research to increase the sample size in clinical studies and validate the role of the SHP2/ERK5 pathway in an in vivo asthma model. Secondly, this study explored only the SHP2/ERK5 pathway in the regulatory mechanism of neutrophilic asthma. However, it was not determined whether it also applies to type 2 inflammation and eosinophilic asthma. Therefore, addressing these issues will help to improve the credibility of this study.
In conclusion, our study demonstrated that increased SHP2 expression level was associated with clinical severity in patients with asthma. Moreover, the SHP2/ERK5 pathway was involved in the regulation of neutrophil auto- phagy and the formation of NETs. Therefore, SHP2 has the potential to become a new indicator for determining the severity and treatment of asthma.