






Hypocomplementemic urticarial vasculitis syndrome (HUVS) is a rare leukocytoclastic vasculitis characterized by recurrent urticarial lesions and hypocomplementemia, often associated with other systemic symptoms such as fever, arthralgias, arthritis, abdominal pain as well as ocular, renal, pulmonary and gastrointestinal involvement. It is the most severe form of three distinct urticarial vasculitis: normocomplementemic urticarial vasculitis, hypocomplementemic urticarial vasculitis (limited to the skin or secondary to a systemic inflammatory disease) and hypocomplementemic urticarial vasculitis syndrome (McDuffie syndrome). The latter is considered by some to be a condition independent of other systemic inflammatory diseases as most cases are idiopathic, yet the syndrome may occur in association with autoimmune diseases, drug reactions, infections or malignancy [1, 2]. Skin findings of this disease can be difficult to distinguish from those of other types of urticaria, namely chronic spontaneous urticaria (CSU).
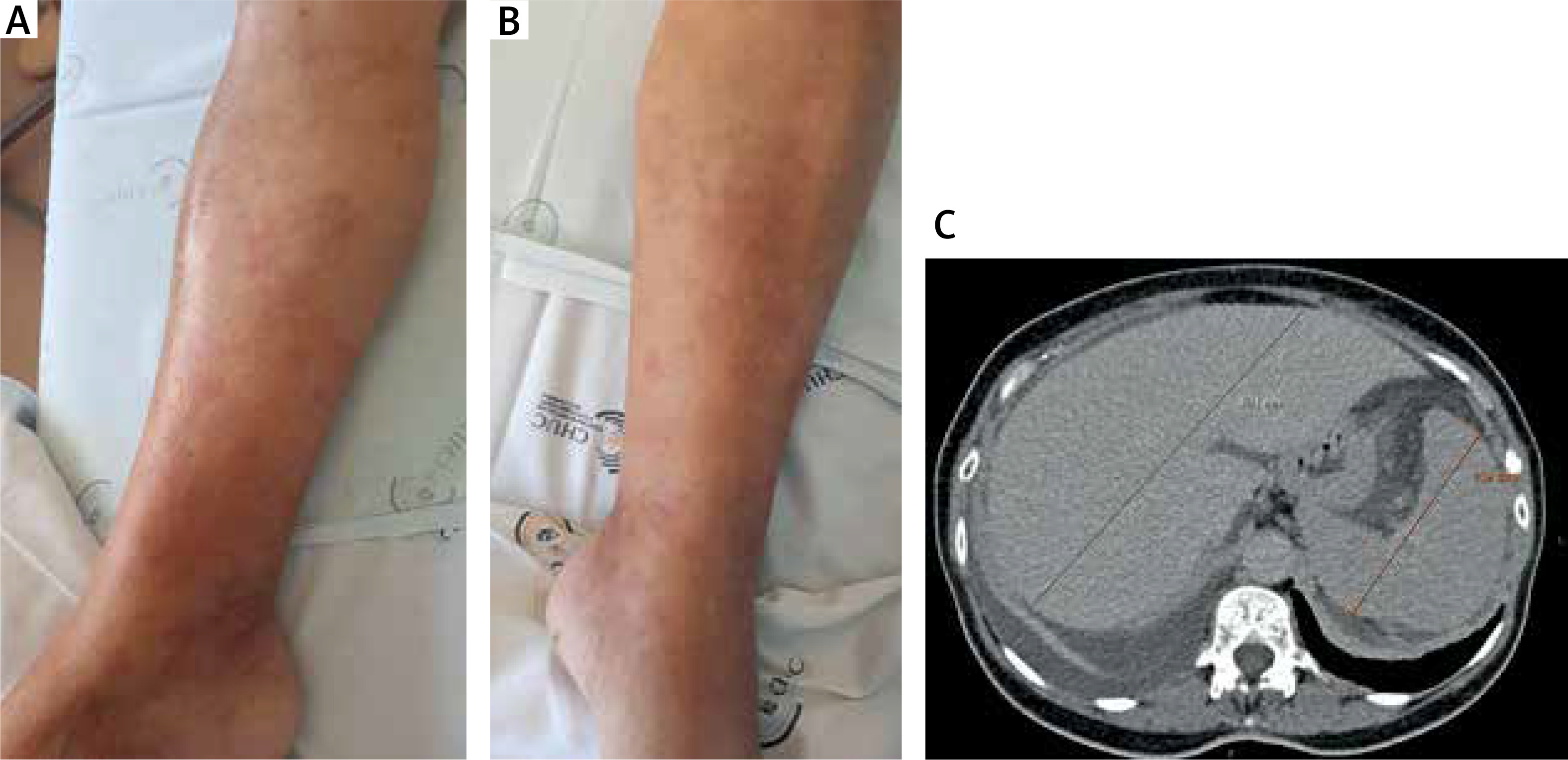
A 70-year-old female presented at the emergency department with sudden onset abdominal pain and distension, fever, anorexia and diarrhoea. She mentioned recurring generalized arthralgias with 4-week evolution as well as recurrent, intensely pruritic urticaria-like lesions lasting more than 24 h, primarily localized in the abdomen and lower extremities, without angioedema, that did not improve with antihistamine therapy, within the previous 6 months (Figures 1 A, B).
Figure 1
Urticarial skin lesions on the right leg (A) and the left leg (B); computed tomography revealing hepatosplenomegaly (C)
The patient had a previous history of well-controlled CSU, diagnosed over 30 years ago, without any skin lesions nor need for medication. Recently she was being closely followed by an Haematology specialist for suspected chronic lymphocytic leukemia.
In laboratory analysis, anemia, elevated erythrocyte sedimentation rate, eosinophilia, decreased complement fragments (C3, C4 and CH50), and impaired kidney function with proteinuria and haematuria were detected (Table 1). Immunological analysis of serum was highly positive for anti-C1q autoantibodies at 67.9 U/ml (normal range: < 10 U/ml) and negative for p-ANCA (perinuclear anti-neutrophil cytoplasmic antibodies), c-ANCA (cytoplasmic ANCA), ANA (antinuclear antibodies) and cryoglobulins. Viral serologies such as hepatitis, Epstein Barr and SARS-CoV2 were all found to be negative.
Table 1
Initial laboratory findings and after 4 weeks of treatment
Imagiological findings revealed generalized lymphadenopathy, hepatosplenomegaly and polyserositis (ascites, pleural and pericardial effusion) (Figure 1 C).
Although skin lesions were initially attributed to CSU, after being evaluated by an allergy specialist, a decision to perform skin biopsy was made, due to some of the atypical characteristics of the skin lesions. Biopsy showed damage of dermal vessels with an infiltrate in the superficial and deep dermis (either perivascular or interstitial dermis), consisting of neutrophils and numerous eosinophils. Distortion of the architecture of multiple capillary vessels and deposition of eosinophilic and basal debris were observed. Eosinophils and basophils on collagen fibres, outlining formation of candle flame figures were also observed suggesting urticarial vasculitis. Therefore, a diagnosis of hypocomplementemic vasculitis syndrome was made in the setting of a paraneoplastic manifestation of the haematological malignancy.
The patient was started on corticosteroid therapy (prednisolone 0.5 mg/kg/day) with progressive clinical improvement of the dermatological findings (asymptomatic residual hyperpigmentation of the skin), absence of fever and other systemic symptoms, as well as laboratory improvement. Treatment was stopped after 4 weeks of therapy with complete regression of the skin lesions and normalized complement levels, haemoglobin and eosinophil count and normal renal function (Table 1).
HUVS is a rare condition with a wide variety of associated manifestations, therefore its pathophysiology is not yet completely understood, although it is considered to be mediated by antigen-antibody complexes that are deposited on vascular lumina, resulting in complement activation through the classical pathway [1, 2]. The association between HUVS and malignancies, particularly Hodgkin’s and non-Hodgkin’s lymphomas has been reported, yet the mechanism behind this association is not fully established, as to whether hypocomplementemic urticarial vasculitis is the cause or the consequence of the malignancy [3].
It is important to be able to differentiate skin lesions related to urticarial vasculitis from those arising from chronic urticaria, as the former last more than 24 h, are sometimes described as painful or burning rather than pruritic and may cause residual hyperpigmentation of the skin [1, 4, 5], yet it should be taken into account that 5% to 10% of patients with CSU may present with urticarial vasculitis [2]. Also, it is crucial to acknowledge that CSU is more responsive to antihistamine therapy [4], whilst urtcarial vasculitis usually responds to corticosteroids, leading to remission of more than 80% of skin lesions [1, 6].
This report illustrates a rare case of hypocomplementemic urticarial vasculitis syndrome as part of a paraneoplastic manifestation following the diagnosis of chronic lymphocytic leukemia. Furthermore, it should be noted that skin lesions may develop even in patients with a prior history of chronic spontaneous urticaria. The presence of atypical lesions, lasting more than 24 h, associated with absence of clinical response to antihistamine therapy should arise one’s clinical suspicion towards this pathology. Skin biopsy is essential for an accurate diagnosis.