

Introduction
Mesenchymal stem cells (MSCs) are specialized cells with therapeutic potential for many diseases, including systemic diseases, because of their ability to migrate into damaged tissue, immune modulation, anti-tumorigenicity, trigger vascularization, and promote epithelial-mesenchymal transition and regeneration [1, 2].
Mesenchymal stem cells can be isolated from bone marrow, adipose tissue, menstrual blood, endometrial polyps, umbilical cord (UC), and other tissues. Two waste tissues that are good suppliers of MSCs are umbilical cord and Wharton’s jelly. Wharton’s jelly derived mesenchymal stem cells (WJ-MSCs) exhibit immunomodulatory properties, strong homing abilities and differentiation and regeneration capabilities. Owing to their fascinating stem cell properties and therapeutic potential WJ-MSCs hold promise in many aspects of regenerative medicine [3, 4]. UC and WJ-MSCs are used in the treatment of diseases such as graft versus host disease (GVHD) that develops after hematopoietic stem cell transplantations and COVID-19 pneumonia, which can lead to an excessive immune response due to their immune-regulating effects [5, 6].
The common term for membrane-containing particles that are released spontaneously from cells but lack a functioning nucleus is extracellular vesicles (EVs). EVs contain many different molecules from inside the cell. These are various proteins (receptors and their ligands, cytokines, etc.), nucleic acids (DNA, RNA, miRNA, etc.) and lipids. According to their vesicle size and biogenesis mechanism, EVs are typically divided into three subtypes: apoptotic bodies (50-5000 nm diameter), microvesicles (100-1000 nm diameter), and exosomes (50-200 nm diameter) [7-9]. Apoptotic bodies are formed by cell lysis during late stages of apoptosis. EVs can be identified by the detection of DNA and histones [10]. Microvesicles (MVs) are formed via direct outward budding of the plasma membrane. They are released into the extracellular environment after selective incorporation of proteins, nucleic acids and lipids and are more heterogeneous than exosomes. There are no specific signs for identifying MVs [11]. Exosomes are formed after the fusion of multivesicular bodies (MVB), a type of late endosomes in the endolysosomal pathway, with the plasma membrane. They have a bilayer membrane. All cell types secrete them, and they are present in the majority of bodily fluids, such as saliva, urine, and blood. Exosomes are composed of heat shock proteins, transport proteins and various lipids such as ceramides, phosphoglycerides, cholesterol, sphingolipids and saturated fatty acid chains [12-16]. The bilayer lipid membrane structures protect exosomes from degradation in the extracellular environment, thus enabling the delivery of cargoes such as mRNA, non-coding RNA such as micro RNAs (miRNAs), long non-coding RNAs (lncRNAs), etc. for the rapid transmission of gene expression in the target cell [17, 18]. These vesicular structures released from stem cells, cancer cells, and many other cells into the extracellular space are responsible for cellular communication and are nanobiologically suitable carriers owing to their low immunity and toxicity [19].
Inflammation is an immune response to a specific stimulus. It is mostly caused by various infections and tissue damage, aids in tissue healing, and is considered a defense mechanism. By facilitating the release of inflammatory mediators, activated M1 macrophages drive the histopathology of chronic inflammation and enhance the inflammatory response [20]. The multiple functions of macrophages in angiogenesis, wound healing, and tissue development have attracted significant interest from researchers over the past few years. The direct role of macrophages in potentially harmful inflammatory processes should not be disregarded when discussing the complex biology of macrophages. When danger signals are received, macrophages quickly release a significant quantity of inflammatory cytokines [21].
Antigen-delivering macrophages and progenitor monocytes, which are white blood cells, mediate the innate immune responses, inflammatory processes, and adaptive immunity. Monocytes in the bloodstream reach the site of infection or inflammation, where they differentiate into macrophages. These cells are essential for immune system performance, tissue remodeling, and tissue repair. During differentiation, cells lose their ability to proliferate and their antibacterial properties are significantly increased, allowing them to participate in inflammatory and immune responses. These cells undergo a phenotypically dynamic transition, depending on their niche and conditions [22]. The pro-inflammatory or M1-like phenotype and anti-inflammatory or M2-like phenotype are two of the best-characterized macrophage phenotypes. M1 macrophages are linked to the initial phases of inflammation and generate elevated amounts of pro-inflammatory cytokines, whereas M2 macrophages generate reduced levels of pro-inflammatory cytokines and are associated with the resolution of inflammation and the process of tissue repair [23]. M1 is classically activated, whereas M2 is activated alternatively. M1 macrophages are induced by cytokines such as interferon γ (IFN-γ), secreted by helper T lymphocytes type 1 (Th1), and bacterial lipopolysaccharides (LPS). High quantities of CD80, CD86, and CD16/32 are produced by M1 macrophages along with pro-inflammatory cytokines such as interleukin (IL)-12, IL-23, IL-6, and tumor necrosis factor α (TNF-α). In contrast, cytokines that stimulate helper T-lymphocyte type 2 (Th2) cells, including IL-4, activate M2 macrophages. The expression of arginase-1 (Arg-1), CD206, IL-10 and chemokines CCL17 and CCL22 were elevated in M2 macrophages [24, 25].
The U937 cell line is a non-adherent cell line derived from histiocytic lymphoma. These cells differentiated into macrophages and dendritic cells. Under certain stimuli, macrophages exhibit physiological and morphological characteristics [26]. Various protocols have been tested for macrophage differentiation in U937 cells [27-30]. Among these, procedures employing phorbol-12-myristate 13-acetate (PMA) have been shown to produce significantly superior outcomes than other documented protocols in terms of cell shape, macrophage surface marker expression, and cytokine production [31]. It has been shown that U937 cells, which we used in our study, transform into a macrophage phenotype as a result of stimulation by expressing CD11c, a macrophage-specific surface antigen [32]. LPS is an outer membrane component of gram-negative bacteria that plays an important role in the pathogenesis of septic shock and various inflammatory diseases by causing overproduction of inflammatory cytokines [33]. The inflammation-inducing effect of LPS occurs when LPS binds to CD14 on the macrophage membranes. Consequently, the NF-κB pathway is activated, leading to increased production of inflammatory factors and nitric oxide (NO), thereby inducing inflammation [34].
Mesenchymal stem cells secrete a wide range of immune regulatory factors that direct macrophages [35]. Another effect of macrophage polarization is the shift in the ratio of pro- to anti-inflammatory factors. The M1 and M2 macrophage phenotypes are among the main factors that shape the course of inflammation. One of the main mediators that play a crucial role in regulating the immune system is the secretion of EVs. These cells act as orchestra conductors in directing the immune system by secreting EVs with different contents according to their environmental conditions. Under culture conditions, cells are cultured without the effects of the microenvironment. It is thought that WJ-MSCs can show their repair and/or regulatory effects by secreting the necessary proteins according to the requirements of the conditions in which they are found in the living environment [36]. In addition, there are many studies on the allogeneic use of EVs, which are active in intercellular communication, in many diseases without stimulating immunity [37]. In our study, we investigated the effectiveness of EVs secreted by WJ-MSCs, which are thought to have immune regulatory activity in the inflammation environment, when stimulated with pro-inflammatory cytokines under culture conditions. We examined the effects of EVs, obtained with or without stimulation of inflammatory cytokines, on U937 cells transformed into the M1 macrophage phenotype induced by PMA and LPS.
Material and methods
Experimental design
The laboratories of Kocaeli University Stem Cell and Gene Therapy Research and Application Center completed the experimental procedures for this study. In this study, experimental groups were designed to examine the effects of extracellular vesicles obtained without stimulation with inflammatory cytokines and those obtained by stimulation with inflammatory cytokines on U937 macrophages. The U937 cell line is a monocytic leukemia cell line that propagates in suspension. To understand the changes caused by EVs in macrophages during inflammation, U937 cells stimulated with PMA and converted into macrophages were used as the control group. Phorbol-12-myristate 13-acetate (PMA) was added at a concentration of 250 ng/ml to induce cell differentiation into macrophages. The cells were then incubated at 37°C in 5% humidified CO2 for 24 h. Following this incubation period, the medium containing PMA was aspirated and LPS was added at a concentration of 10 µg/ml. The apoptotic and antiproliferative effects of EVs and sEVs on U937 macrophages were evaluated in vitro. Experimental groups:
Cell culture of U937 and WJ-MSCs
U937 cells exhibiting monocyte morphology were obtained from ATCC (CRL-1593.2) and WJ-MSCs were obtained from STEMBIO Cord Blood, Cell and Tissue Center, TÜBİTAK Martek, Gebze, Turkey. As the study was conducted using cell lines, ethical approval was not required.
U937 cells were cultured in Roswell Park Memorial Institute (RPMI) medium (Gibco, Paisley, UK) containing 10% fetal bovine serum (FBS) (Gibco), 1% penicillin-streptomycin (Gibco), and 1% GlutaMax (Gibco). U937 cells were seeded in T75 suspension culture flasks with 1 × 106 cells and incubated at 37°C in an incubator with 5% humidified CO2 and 95% air. For the medium exchange of U937 cells in the suspension culture, T75 suspension culture flasks were placed in an upright position, and after waiting for 3 min, 3-4 ml of waste medium was carefully removed from the surface and replaced with fresh medium. Cells were passaged when they reached 70-80% confluence.
Wharton’s jelly mesenchymal stem cells 1 × 106 were seeded in T175 flasks, cultured in DMEM-F12 medium (Capricorn Scientific, Ebsdorfergrund, Germany) containing 10% FBS, 1% penicillin-streptomycin, and 1% Gluta- Max, and incubated at 37°C in an incubator containing 5% humidified CO2. The medium was changed every three days. When the cells were 70-80% confluent, they were passaged and transferred to new culture dishes for expansion.
Wharton’s jelly mesenchymal stem cells (1 × 106 cells) were seeded in T175 culture dish and when they reached 60-70% confluency, the medium containing pro-inflammatory cytokines with IFN-γ, TNFα, and IL-1β 10 ng/ml each (BioLegend, San Diego, CA) was changed and cultured [38, 39]. In this process, the cells were grown in a stimulation medium for 72 h at 37°C with 5% CO2. Afterwards, serum-free medium was added to the culture flask, it was washed twice with phosphate-buffered saline (PBS) (Capricorn) and the medium was collected for EV isolation after 24 h. For EVs from unstimulated WJ-MSCs, no cytokines were added to the medium and all other experimental conditions were identical.
Characterization of WJ-MSCs
The phenotypic characteristics of the cells were evaluated by flow cytometry in terms of mesenchymal stem cell surface markers. In addition, cells were subjected to adipogenic, osteogenic, and chondrogenic differentiation to demonstrate their multipotent properties [40].
Flow cytometry analysis of WJ-MSCs
For flow cytometric analysis, cells were counted after trypsin treatment and transferred into tubes with approximately 0.2 × 106 cells/200 µl medium for each antibody. To each tube, 10 µl of the respective fluorescein isothiocyanate (FITC)-and phycoerythrin (PE)-conjugated monoclonal antibodies CD45, CD90, CD105, HLA-DR, CD73, CD34, CD106, and CD44 (BD Biosciences, California, USA) and the appropriate isotype controls (IgG1 and IgG1/G2a) were added and incubated for 45 min. After incubation, PBS was added and the unbound antibodies were removed by centrifugation at 1800 rpm for 5 min. The cells were analyzed in 400 µl of washing solution in a flow cytometry device (FACSCalibur, BD Biosciences, USA) using the BD CellQuest software.
Differentiation experiments
Wharton’s jelly mesenchymal stem cells (1.5 × 105 cells) were seeded in 6-well culture dishes and standard medium (DMEM-F12 containing 10% FBS and 1% penicillin/streptomycin) was replaced with adipogenic differentiation medium containing DMEM Low Glucose (LG) (Capricorn), 10% FBS, 1% penicillin/streptomycin, 10-6 M dexamethasone, 0.5 mM isobutyl-methylxanthine, 200 µM indomethacin, and 10 g/ml insulin when 50-60% confluency was reached. The medium was refreshed every 2-3 days and fixed samples after 3 weeks of culture were stained with Oil Red O, and microscopic examination revealed cells with accumulated lipid droplets [41].
Wharton’s jelly mesenchymal stem cells that were similarly seeded and the osteogenic differentiation medium that included 100 nM dexamethasone, 0.05 µM ascorbate-2-phosphate, and 10 mM β-glycerophosphate was replaced when 80-90% confluency was reached and the medium was refreshed every 2-3 days. Osteogenic differentiation was also demonstrated by staining calcified nodules with Alizarin Red S [41]. For differentiation, the control groups were cultured for the same time and under the same conditions as the differentiation groups, but standard culture medium containing DMEM-F12 containing 10% FBS and 1% penicillin/streptomycin was used throughout the culture period. Chondrogenic differentiation was performed by micromass culture, and the cells were stained with Alcian blue. For the chondrogenic differentiation, a previously published protocol was used [42].
Images were captured using a light microscope (Olympus IX71, Tokyo, Japan).
Isolation of EVs and sEVs
Extracellular vesicles and sEVs were isolated and characterized from approximately 120-200 ml of medium obtained from at least 6-10 T175 flasks each time using the differential ultracentrifugation technique as described previously [41]. After collecting the MSC culture media, the cells were centrifuged for 10 min at 300 × g, and dead cells were extracted. The supernatant was transferred to fresh tubes and centrifuged at 15,000 × g for 20 min to remove cellular debris, and the supernatant was transferred to fresh tubes. To remove microvesicles larger than 220 nm, the supernatant was filtered through a 0.22 µm pore size filter (BIOFIL, China). A CS150FNX (Hitachi Himac, Japan) ultracentrifuge and an S50A type rotor were used to centrifuge the pre-purified cell medium for 70 min at 4°C and 110,000 g. After removing the supernatant, a small amount of PBS (~100-600 µl) was used to suspend the EV pellet. Protein quantification was performed using a bicinchoninic acid (BCA) kit (Boster, Pleasanton, CA).
Characterization of EVs and sEVs
Flow cytometry analysis of EVs with magnetic beads
Extracellular vesicles and sEVs were incubated with Exosome-Human Flow Detection Reagent (from cell culture) (Invitrogen by Thermo Fisher Scientific, Vilnius, Lithuania). Dynabeads (magnetic beads), typically 2.7 µm in diameter, were coated with antibodies against CD9, CD63 and CD81. Each type of these specific magnetic beads was incubated overnight at 4°C in separate tubes. EV-bead complexes were washed with PBS using a magnet, incubated with fluorophores containing phycoerythrin (PE)-bound anti-CD9, anti-CD63, and anti-CD81 monoclonal antibodies (PE Mouse anti-human, BD Pharmingen) at room temperature for 45 min in the dark, and analyzed using flow cytometry (FACSCalibur, BD Biosciences, USA) [41].
Dynamic light scattering (DLS) analysis
Extracellular vesicles were placed in a clear, four-sided cuvette and diluted to 1 : 50 in PBS. The sizes of the EVs and sEVs were measured using a Zetasizer Nano ZS90 v7.01 (Malvern, UK). Three scattering measurements of size and density were recorded for each sample, and the average size was obtained.
Scanning electron microscopy analysis
An environmental scanning electron microscope (ESEM, Quattro S, Thermo Fisher Scientific, US) was used for morphological characterization of EVs and sEVs. Five microliters of EV samples were dropped and dried onto the grid and analyzed using a STEM 3 detector. The images were taken at an electron beam voltage of 30 kW and a spot size of 1.0.
Investigation of the effects of WJ-MSC-EVs on in vitro inflammation model
In vitro inflammation modeling of U937 cells induced by PMA and LPS
For the transformation of cells into macrophages, 250 ng/ml phorbol-12-myristate 13-acetate (PMA) was added and incubated at 37°C and 5% CO2 for 24 h. The medium of U937 cells that adhered and acquired macrophage morphology after PMA treatment was replaced with medium containing 10 µg/ml LPS (Escherichia coli, serotype O26:B6), and the cells were incubated in medium with LPS at 37°C in an incubator with 5% CO2 for 24 h [43]. After PMA and LPS treatment for one day each, an in vitro inflammation model was established and the efficacy of the model was tested with flow cytometry. CD11b, CD11c, and CD86 markers were analyzed to demonstrate the efficiency of this transformation in U937 cells that were not stimulated, were only stimulated with PMA, and were transformed into the M1 phenotype upon stimulation with PMA + LPS. Cells were labeled with anti-CD11b, anti-CD11c, and anti-CD86 antibodies for 45 min. The cells were then examined using the flow cytometer [44].
Co-culture of U937 macrophages with EVs and sEVs
Isolated EVs and sEVs were co-cultured with U937 cells treated with PMA + LPS. U937 cells without EVs were used as the control groups (P and P + L). The experiments were performed in 6-well culture dishes. Cells were seeded onto glass slides in wells for immunostaining. Each well was incubated for 48 h with 100 ng/ml EVs and sEVs, and the amount of protein was determined by BCA assay. The cells were examined daily under a microscope to observe morphological changes.
Confocal microscopy, flow cytometry, quantitative real-time polymerase chain reaction (qRT-PCR) and Annexin V/PI cell apoptosis analyses were performed to examine the effects of WJ-MSC-derived EVs in an in vitro inflammatory model.
Immunofluorescence staining and confocal microscopic examination
Samples were incubated in the appropriate block serum for 20 min and then incubated with anti-Proliferating Cell Nuclear Antigen (PCNA) (1 : 250, ab92552, Abcam, Cambridge, UK) and anti-TNF-α (1 : 200, NBP2-34372, Novus Biologicals, Colorado, USA) primary antibodies overnight at +4°C. After washing with PBS, the samples were incubated with the appropriate fluorescently labelled secondary antibodies (1 : 1000, ab150083 for PCNA, ab150113 for TNF-α, Abcam) for 1.5 hours at room temperature. Following a washing procedure to eliminate any unbound antibodies, the samples were covered with UltraCruz Mounting Medium for fluorescence with DAPI (4′,6-diamidino-2-phenylindole). A laser scanning confocal microscope (Leica DMI SP8) was used to examine and capture images of the samples. Quantitative values we obtained by immunofluorescent staining using ImageJ software.
Annexin V/PI for detection of apoptosis
The kits were used to identify apoptotic cells that expressed Annexin V/PI (BD Bioscience-FITC Annexin V Apoptosis Detection Kit I, USA), in compliance with the manufacturer’s instructions. Trypsin-EDTA was used to extract cells from the culture dish surface. Next, 3 × 105 cells were rinsed with cold PBS, and the cell pellets were homogenized once more using 100 µl of binding buffer. FITC annexin V and PI were added and the mixture was allowed to sit at room temperature for 20 min. After incubation, 300 µl of binding buffer was added, the reaction was stopped, and the outcomes were examined using a FACSCalibur flow cytometer.
Investigation of gene expression levels by qRT-PCR
Quantitative RT-PCR was performed using primers specific for these genes (Table 1) to observe changes in IFN-γ, IL-6, IL-10, IL-1β, and TNF-α gene expression in U937 macrophages in all experimental groups. Total RNA was isolated from the samples obtained from the experimental groups using a PureLink RNA Mini Kit (Thermo Fisher Scientific, CA, USA) following the manufacturer’s instructions. cDNA synthesis was performed from the directions included with a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Vilnius, Lithuania). The samples of synthesized cDNA were stored at –20°C until the experiment was set up. qRT-PCR was performed using a real-time PCR device (Roche LC480II; Mannheim, Germany). Table 1 contains a list of the primers and base sequences for the genes used in the experimental groups. For each sample, a 20 µl reaction mix containing RealQ Plus 2x Master Mix Green (Ampliqon, Denmark) SYBR dye was prepared. PCR conditions were incubation at 95°C for 2 min, followed by 45 cycles of 95°C for 5 s and 60°C for 60 s. The Cp results were analyzed using the 2-ΔΔCT method relative to the reference gene (ACTβ) and control group.
Table 1
Base sequences of primers used for real-time PCR
Statistical analysis
Data were analyzed using the statistical software SPSS (IBM Corp., version 20.0;, Chicago, IL, USA). Each experiment was independently replicated at least three times, and within each experiment, three or more biological replicates were included. Numerical variables are presented as mean ± standard error of the mean (SEM). The paired t-test and Kruskal-Wallis one-way ANOVA test were used to evaluate the differences between the groups. *p < 0.05 indicated a significant difference between the experimental and control groups; **p < 0.01 and ***p < 0.001 indicated a highly significant difference.
Results
Characterization of WJ-MSCs
When the WJ-MSCs reached the third passage, they were analyzed using flow cytometry. CD105, CD90, CD73, and CD44 levels were over 90% positive, whereas the hematopoietic markers CD45, CD34, CD106, and HLA-DR were lower than 1% (Fig. 1A).
Fig. 1
Characterization of Wharton’s jelly mesenchymal stem cells (WJ-MSCs): A) flow cytometry analysis of cells B) osteogenic differentiation control, C) osteogenic differentiated group, D) adipogenic differentiation control, E) adipogenic differentiated group, F) chondrogenic differentiation not stained, G) and chondrogenic differentiated group stained with alcian blue. Scale bar: 50 µm
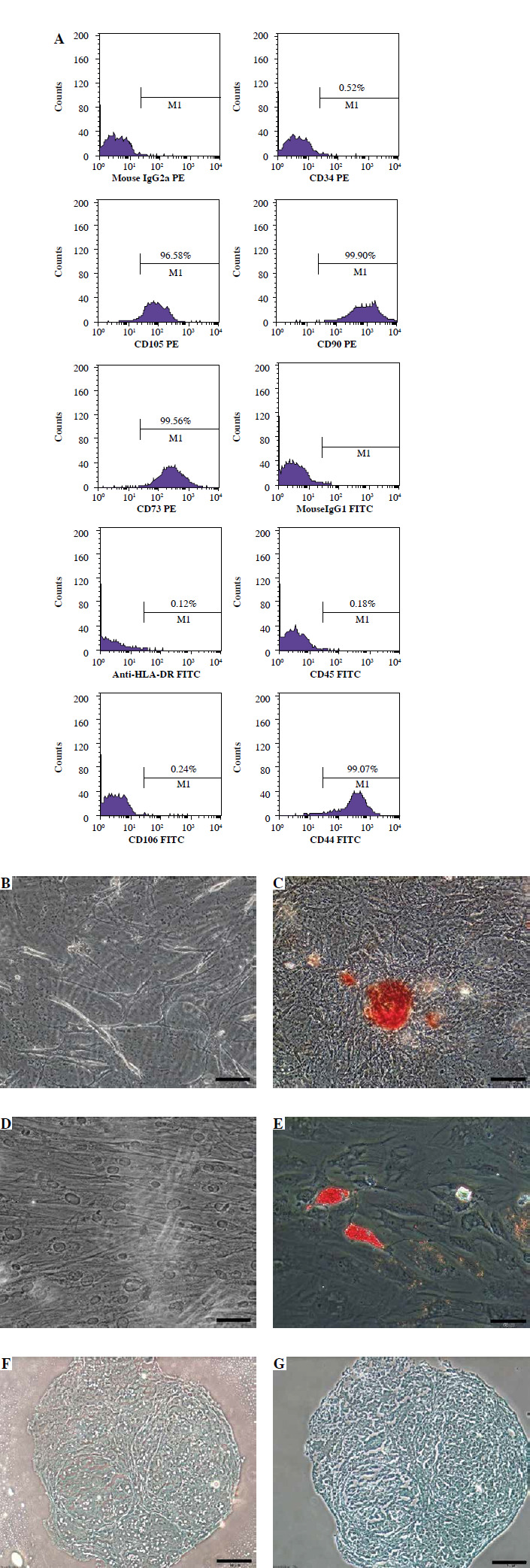
Wharton’s jelly mesenchymal stem cells were incubated separately in adipogenic and osteogenic differentiation media for three weeks, after which the cells were fixed. The osteogenic and adipogenic differentiation groups were stained with Alizarin Red S and Oil Red O, and alcian blue, respectively (Fig. 1C, E, G). No staining was observed in the control groups (Fig. 1C, D, F). The results were visualized under a light microscope (Olympus).
Phase-contrast microscopy showed that WJ-MSCs stimulated with inflammatory cytokines were more fusiform than those that were not stimulated (Fig. 2A, B).
Characterization of EVs and sEVs
Extracellular vesicles used for characterization were isolated from approximately 120 ml of the medium obtained from six T175 flasks. EV and sEV protein concentrations obtained from this amount of medium were found to be 45.33 ±4.93 µg/ml and 51.67 ±8.32 µg/ml using the BCA kit.
Flow cytometry analyses showed that sEVs had slightly lower positivity for CD63 (EVs 91.47 ±0.85%/sEVs 84.87 ±3.87%) and CD81 (EVs 94.67 ±3.68%/sEVs 91.42 ±3.20%) surface markers and much lower positivity for CD9 compared to EVs (EVs 90.73 ±3.22%/sEVs 28.86 ±3.52%) (Fig. 3A). There was no significant difference in the characterization data of stimulated EVs compared with unstimulated ones except for CD9.
Fig. 3
Characterization of extracellular vesicles (EVs) and stimulated sEVs. A) Flow cytometric dot blot analysis of EVs with anti-CD9, anti-CD63 and anti-CD81 surface markers (upper left quadrant [UL] indicates positive cells); B) Size distribution of EVs and sEVs determined by dynamic light scattering (DLS) C) Size and shape analysis of EVs by electron microscopic examination; D) Mean concentration/size graphs of EVs and sEVs were assessed by nanoparticle tracking analysis (NTA)
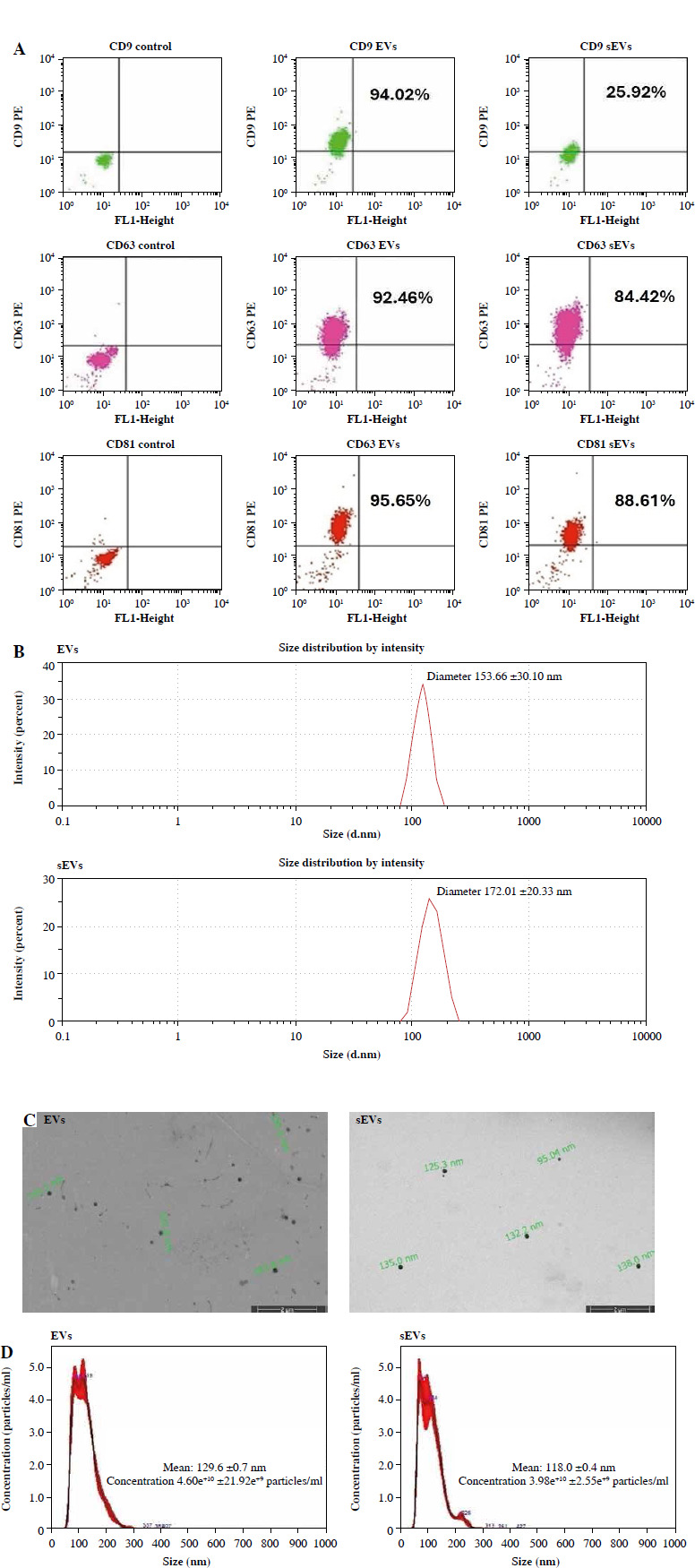
Three scattering measurements for size and density were recorded for each sample during dynamic light scattering (DLS) measurements. The EVs had a diameter size of 153.66 ±30.10 nm and for sEVs it was 172.01 ±20.33 nm (Fig. 3B).
Scanning electron microscopy (SEM) imaging revealed that EVs had a round morphology. The average size of EVs was 148.66 ±31.01 nm and that of sEVs was 121.0 ±17.29 nm (Fig. 3C).
Nanoparticle tracking analysis (NTA) results (EVs 129.6 ±0.7 nm, sEVs 118.0 ±0.4) confirm DLS and SEM analysis. Particle quantity per ml was 4.60e+10 ±21.92e+9 particles/ml for EVs, while for sEVs it was 3.98e+10 ±2.55e+9 particles/ml.
Establishment of in vitro inflammation model by inducing U937 cells with PMA and LPS
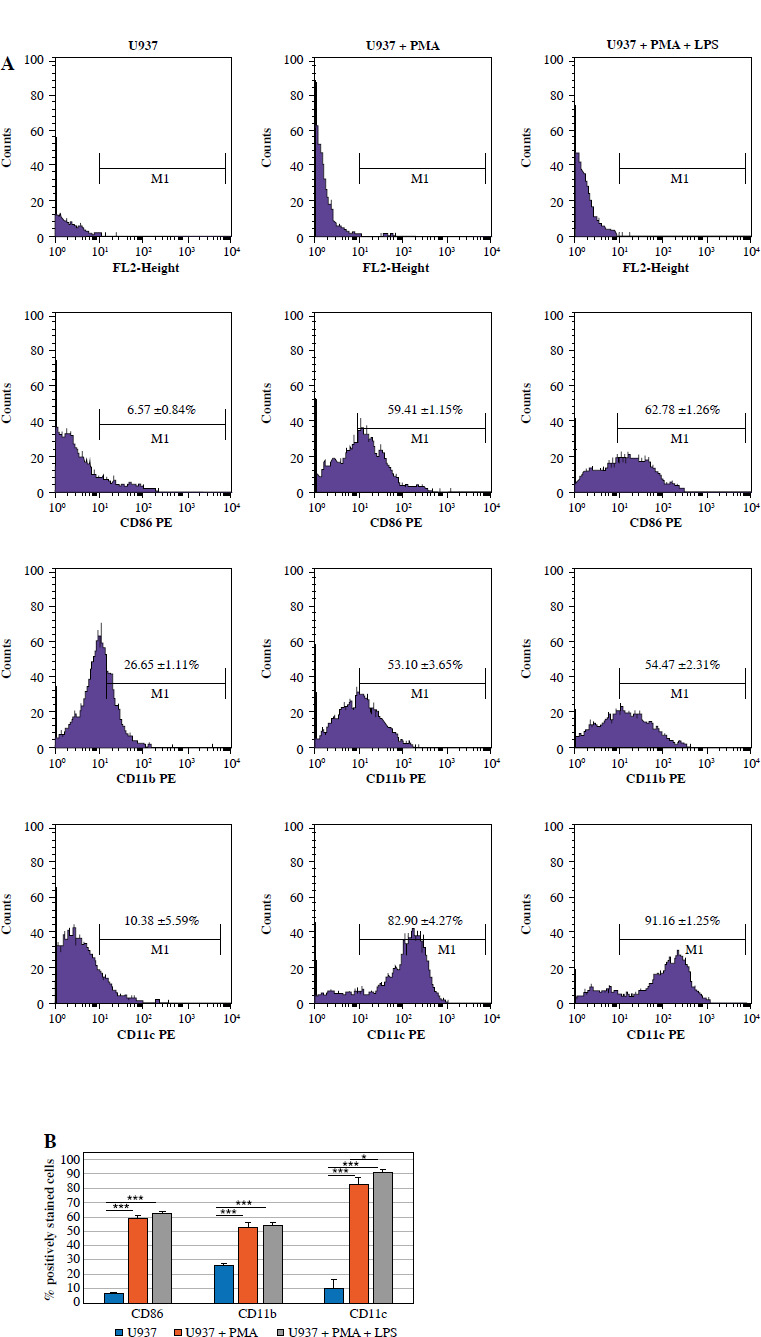
CD86, CD11b, and CD11c markers were immunolabelled in U937 cells, U937 cells stimulated with PMA (P) alone, and U937 cells transformed to the M1 phenotype upon stimulation with both PMA and LPS (P + L). The efficiency of this transformation was graphically demonstrated using flow cytometry (Fig. 4). The percentage of CD86, CD11b and CD11c positively stained cells was significantly higher (p < 0.001) after P and P + L stimulation compared to that in U937 cells. The relative increases in P and P + L were significant for CD11c (p < 0.05).
Fig. 4
A) Confirmation of induced U937 macrophage differentiation by flow cytometry. CD86, CD11b and CD11c examination of U937 cells, U937 stimulated with PMA only and U937 cells stimulated with both PMA and LPS B) The data represent the mean ± SEM of % positively stained cells (n = 3). * indicates statistical significance level (*p < 0.05, ***p < 0.001)
Investigation of the effects of EVs and sEVs on an in vitro model of inflammation
To examine the effects of EVs obtained from WJ-MSCs on pro-inflammatory cytokine release in an in vitro inflammation model, samples were labelled with TNF-α antibody, and the effects on proliferation were examined using PCNA antibody (Fig. 5).
Fig. 5
Immunofluorescence staining and confocal microscopic examination of U937 cells. Experimental groups containing: A) PMA (P), B) PMA + LPS (P + L), C) PMA-LPS-extracellular vesicles (P + L + EVs) and D) PMA-LPS-stimulated EVs (P + L + sEVs). Cell nuclei were labeled with DAPI (blue) (a1-d1), TNF was labeled with Alexa flour 488 (green) (a2-d2), PCNA was labeled with Alexa flour 647 (red) (a3-d3) and images were superimposed (merge) (a4-d4). Measurement bar: 100 μm. Bar graph of immunofluorescence (IF) score of images with ImageJ software (E). The analysis was performed with five biological replicates and analysis of five fields per replicate; * indicates statistical significance level (*p < 0.05, **p < 0.01)

Tumor necrosis factor and PCNA expression between the groups was evaluated using ImageJ. A significantly lower value (*p < 0.05) was observed in TNF expression after culturing with sEVs compared to that in the EVs group. In addition, TNF expression was significantly higher in the EV group than in the PMA and PMA + LPS groups (**p < 0.01 and *p < 0.05, respectively). For the PCNA marker, positivity was higher in the sEVs group than in the EVs co-cultured and PMA group (p < 0.05).
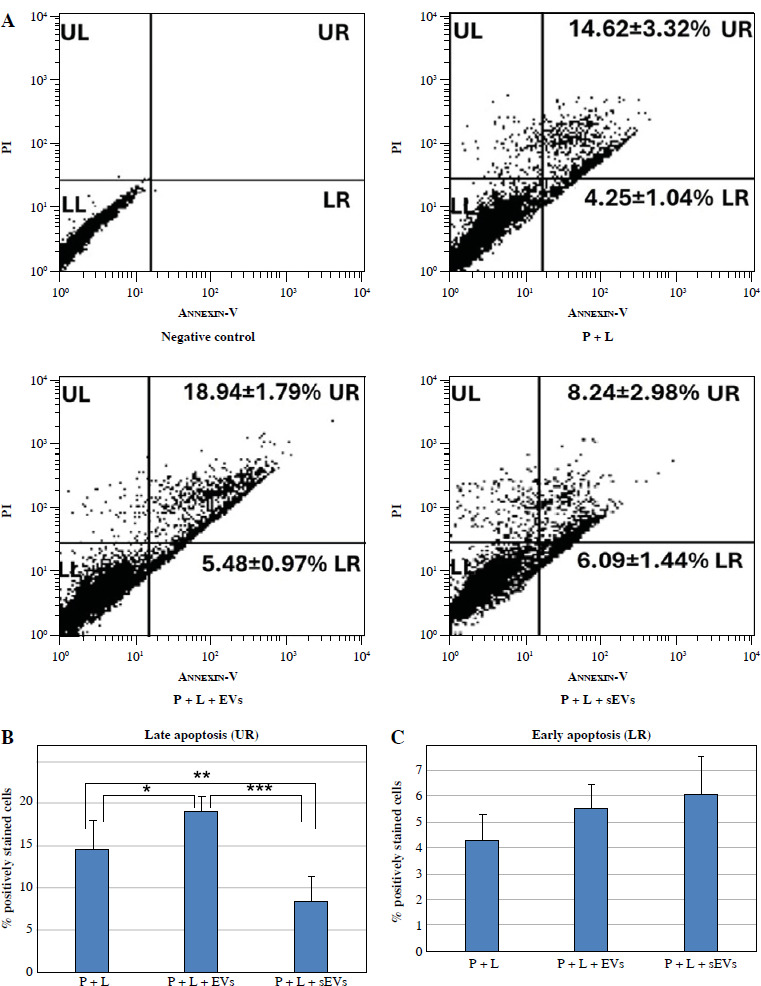
Apoptosis detection by Annexin V/PI labeling
Annexin V-FITC and PI labeling was plotted and calculated from experiments using flow cytometry. Late apoptosis was significantly higher in the P + L + EVs group (*p < 0.05) and significantly lower in the P + L + sEVs group (**p < 0.01) compared to the P + L group. The P + L + sEVs group showed a significantly lower level compared with the P + L + EVs group (***p < 0.001) (Fig. 6B). However, no significant difference was detected in early apoptosis (Fig. 6C).
Fig. 6
Apoptosis/necrosis determination by flow cytometry using Annexin V/PI. A) Scatter plot of flow cytometry analysis, upper left (UL) indicates necrotic or dead cells (Annexin V negative and PI positive), upper right quadrant (UR) late apoptosis (both Annexin V and PI positivity), lower left (LL) represents living cells (Annexin V, PI negative) and lower right (LR) early apoptotic cells (Annexin V positive, PI negative). The experiment was repeated three times and data represent the average of the (B) early apoptotic and (C) late apoptotic cells. Each experiment was performed with three biological replicates (n = 3). * indicates statistical significance level (*p < 0.05, **p < 0.01, ***p < 0.001)
Examination of gene expression levels by qRT-PCR
Changes in pro- and anti-inflammatory markers at the gene level in the cells were analyzed (Fig. 7).
Fig. 7
qRT-PCR analysis of gene expression of inflammation related cytokines in U937 cells. X-axis shows experimental groups, y-axis shows fold changes compared to reference gene. Each experiment was performed with three biological replicates (n = 3). * indicates statistical significance level (*p < 0.05, **p < 0.01, ***p < 0.001)
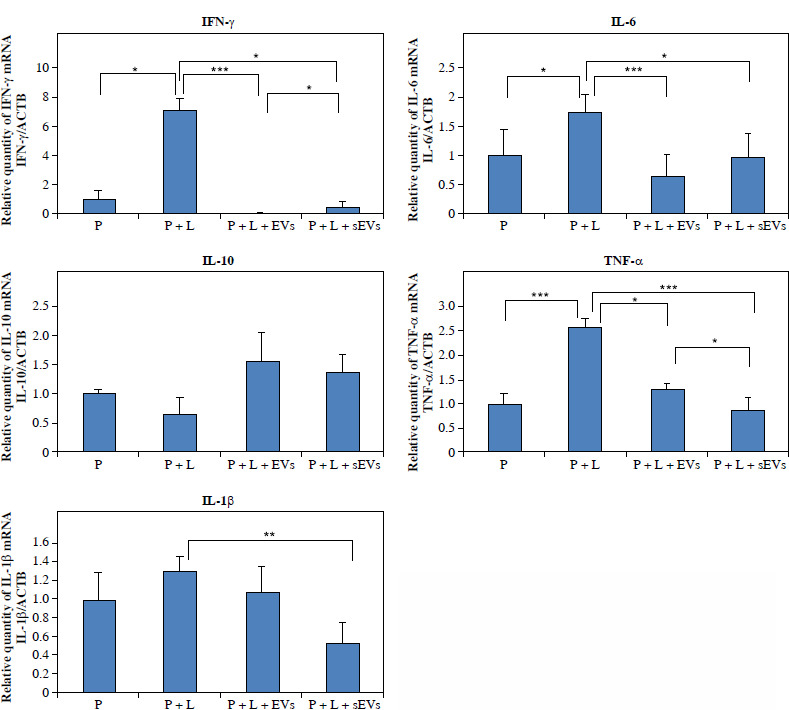
The gene expression of all pro-inflammatory markers was higher in the P + L group compared to that in the P group. TNF-α (***p < 0.001), IFN-γ, and IL-6 (*p < 0.05) showed significant differences, whereas the increase in IL-1β gene expression was not significant. In both the P + L + EVs and P + L + sEVs groups, pro-inflammatory markers IFN-γ, IL-6, TNF-α, and IL-1β (except the EV group) were significantly lower compared to the P + L group (*p < 0.05, **p < 0.01, ***p < 0.001). The higher gene expression of the anti-inflammatory marker IL-10 compared to the P + L group was not statistically significant. When the P + L + EVs and P + L + sEVs groups were compared, a significantly higher value in the latter was observed for IFN-γ (*p < 0.05) and a lower value for TNF-α (*p < 0.05).
Discussion
Extracellular vesicles (EVs) are membrane-enclosed particles that are released from almost every cell and are important for intercellular communication. EVs have several advantages that make them more suitable for use as therapeutic agents. These advantages include the fact that they cannot self-replicate, have a very low risk of ectopic differentiation and tumor formation, and are not perceived as foreign by the cells of the immune system. The ability of MSC-derived EVs (MSC-EVs) to go directly to the site of injury and their immunomodulatory properties appear to be similar therapeutic benefits to those of their parent cells [44-46]. Mesenchymal stem cells are used in the treatment of acute graft versus host disease (aGVHD) in steroid-resistant cases due to their immunomodulatory effects. Interestingly, human bone marrow MSC-derived extracellular vesicles (hBM-MSC-EVs) exhibit many of the hBM-MSC immunoregulatory properties due to their paracrine factor content, which varies greatly according to the collection method. Dal Collo et al. demonstrated the immunological characterization of hBM-MSC-EVs and their ability to induce regulatory T cells (T-regs) both in vitro and in a xenograft mouse model of aGvHD [47].
Conditioned media made from MSCs contain paracrine secretory components that replicate the actions of the entire cell. EVs, growth factors, and cytokines are among the secreted components, known as the secretome. According to recent research, MSC-derived EVs can either decrease M1 and/or increase M2 polarization, depending on the environment [48]. Owing to their immunomodulatory abilities, MSCs have been used in various clinical procedures that require immune response regulation. When administered for an appropriate duration and in appropriate amounts, cytokines stimulate the cells to express more immunoregulatory molecules, facilitating the acquisition of EVs, which are abundant in these molecules. The development of conditioning regimes to obtain functional MSCs or EVs under various pathophysiological conditions will be facilitated using specific in vitro activation parameters.
The human histiocytic lymphoma-derived U937 leukemia cell line and the monocytic cell line THP-1 have been widely used as in vitro models to study the mechanisms of immune modulation [32, 35]. Various protocols have been used to differentiate U937 and THP-1 cells into macrophages [28-31]. To create an inflammation model, we induced U937 cells to acquire an M1 macrophage phenotype. The transformation process into M1 macrophages was completed by first culturing with PMA for one day and then culturing with LPS. The conversion of U937 cells into macrophages was demonstrated by the expression of the macrophage-specific antigens CD86 [49, 50], CD11b [51, 52], and CD11c [33, 53]. Using flow cytometry, we observed increased expression of CD11b, CD11c, and CD86 after macrophage stimulation with PMA in U937 cells. These results, which are consistent with those in the literature, indicate that the stimulation process was successful. In addition, for M1 macrophage stimulation, slightly higher CD86 and CD11c (p < 0.05) expression was observed after incubation with LPS compared to stimulation with PMA alone, while no difference was observed in CD11b expression. Furthermore, qRT-PCR results showed an increase in all pro-inflammatory markers at the gene expression level after LPS stimulation. These increases were significant for IFN-γ (*p < 0.05), IL-6 (*p < 0.05), and TNF-α (***p < 0.001).
Owing to their immunoregulatory abilities, MSCs are used in a variety of clinical procedures that require regulation of the immune response. When administered for an appropriate duration and in appropriate amounts, cytokines stimulate the cells to express more immunoregulatory molecules, facilitating the acquisition of EVs, which are abundant in these molecules. The development of conditioning regimens to obtain functional MSCs or EVs under various pathophysiological conditions is facilitated by the use of specific in vitro activation parameters [48].
In our study, we found that CD9 expression was significantly lower in WJ-MSC-derived sEVs compared to that in EVs. Brosseau et al. published a review showing CD9 expression in hematopoietic cells and the cellular processes involved in the regulation of inflammation and its role in many diseases [54]. Decreased CD9 expression has been found to decrease the ability of human eosinophils to stimulate CD4(+) T cell activation, proliferation, and cytokine production [55]. Similarly, a study reported that CD9 expression in primary cultures of murine peritoneal macrophages was downregulated by IFN-γ [56]. Our study suggested that LPS-stimulated U937 cells co-cultured with sEVs may regulate inflammatory cytokine release; however, further analyses are needed to confirm this supposition.
Based on the hypothesis that EVs secreted by WJ-MSCs in an inflammatory environment (when stimulated) would show better immunoregulatory effects, we examined the changes in inflammatory and apoptotic markers after co-culture of EVs derived from WJ-MSCs cultured with pro-inflammatory cytokines and U937 cells induced to differentiate into M1 macrophages. Our findings showed that WJ-MSC EVs cultured with pro-inflammatory cytokines effectively reduced inflammation. Following co-culture with EVs, the gene expression levels of pro-inflammatory markers were reduced. This decrease was particularly significant for IFN-γ, IL-6, TNF-α and IL-1β in EV co-culture. A non-significant increase in IL-10 anti-inflammatory marker levels was observed after co-culture with both EVs. In our study, TNF-α was examined by confocal microscopy, and it was observed that the EVs of MSCs stimulated with pro-inflammatory cytokines decreased the level of TNF secreted from macrophages of the M1 phenotype, whereas when EVs were added to the PMA-LPS group, proliferation (PCNA) increased in U937 cells. This suggests that MSCs grown in an inflammatory environment (cultured with pro-inflammatory cytokines) may be more effective in regulating inflammation.
Prasanna et al. compared the changes in mesenchymal markers and immunoregulatory properties by culturing BM-MSCs and WJ-MSCs with IFN-γ and TNF-α. For this purpose, they proliferated peripheral blood mononuclear cells (PBMCs) with phytohemagglutinin (PHA) and mitomycin C and observed that WJ-MSCs stimulated after co-culture with mesenchymal cells stimulated or not stimulated with pro-inflammatory cytokines reduced the proliferation of PBMCs [57]. In another study, preconditioning with inflammatory cytokines was found to enhance mesenchymal stem cell properties, including differentiation and immune modulation. Preconditioning with interleukin 1b (IL-1β) and interferon γ (IFN-γ) was performed to increase the immunomodulatory ability of human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSCs), and there was a statistically significant decrease in peripheral blood mononuclear cell proliferation while their immunosuppressive ability increased [58].
López-García et al. cultured BM-MSCs from mice with pro-inflammatory cytokines and transformed M0 macrophages into M1 macrophages using LPS and IFN. Indirect co-culture was performed and preconditioned MSCs were observed to convert macrophages to an anti- inflammatory phenotype [36]. Confocal microscopy images showed a significant decrease in TNF expression in cells, especially after co-culture with stimulated EVs. Furthermore, a significant decrease in pro-inflammatory cytokine levels was observed at the gene level after co-culture with sEVs. In addition, sEVs were found to increase macrophage proliferation compared to EVs. Late apoptosis was also lower in macrophages stimulated after culture with sEVs. According to our results, when WJ-MSCs were conditioned with inflammatory cytokines, the sEV content increased the anti-inflammatory effect on activated (M1) U937 macrophages and increased the proliferative effect. The results showed that WJ-MSC EV content varies according to the conditions in which the cells are present [59] and that the effect on inflammation may vary according to this content. Compared with the EVs of unstimulated MSCs, the sEVs of MSCs cultured in stimulation medium with inflammatory cytokines appeared to contribute to the redirection of macrophages with a pro-inflammatory (M1) phenotype to the anti-inflammatory (M2) phase.
In conclusion, EVs stimulated with proinflammatory cytokines exhibit enhanced anti-inflammatory and regenerative properties. sEVs had a greater apoptotic effect on macrophages from the U937 cell line and reduced inflammatory cytokine release compared to EVs from non-stimulated MSCs. Our study suggests that mesenchymal stem cell EVs, especially those stimulated by pro-inflammatory cytokines, are important cellular components that can be used to alter macrophages from M1 to M2. These effects are mediated by the altered cargo of EVs and their effects on key cellular signaling pathways. Our findings should be supported by additional experiments that show changes in protein level. The clinical potential of EVs for treating inflammatory and degenerative conditions is vast and promising.