






Introduction
Lipomas are the most common soft tissue tumours and occur in about 1% of the population. Physicians evaluate subcutaneous lipomas by palpation, imaging studies, and pathomorphological examination. An additional tool that is gaining in value is genetic testing, which can help in making the correct diagnosis in complicated cases or in differentiating ambiguous lesions. In this review, we focus primarily on the genetic background of lipomas and on the diseases and genetic syndromes in which they coexist. Our aim is to sensitize readers to the fact that even a common lesion may prove to be part of a more complex, rare problem.
Classification and pathomorphology
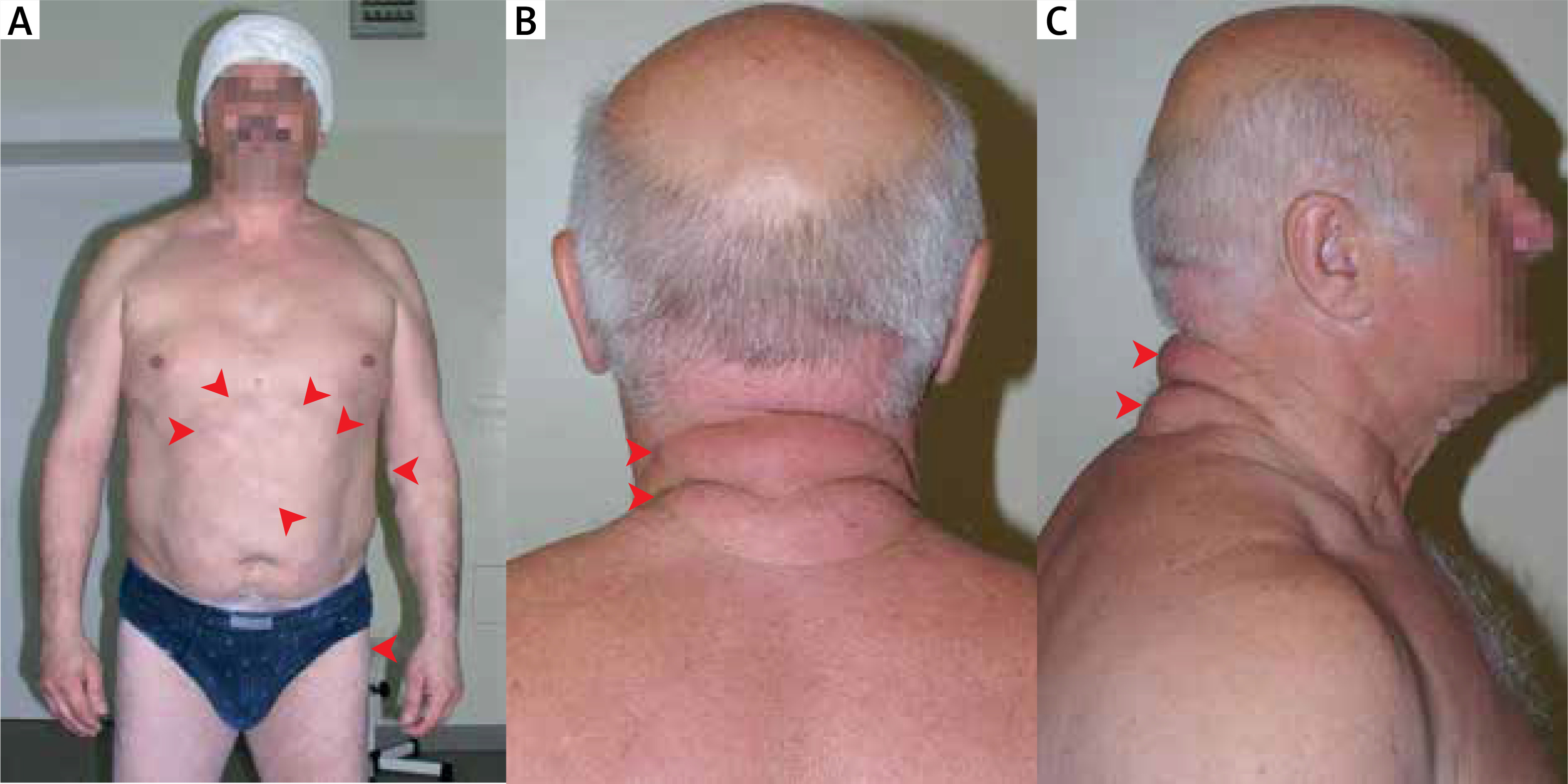
Lipomas belong to benign lipomatous neoplasms and are coded 2E80.0 according to the International Classification of Diseases 11th Revision effective of 1 January 2022. Within this group, lipomas were divided based on their location: subcutaneous (Figure 1 A), subfascial, deep internal or visceral, other specified sites, and unspecified sites. Lipomas are composed of mature white adipose tissue. They are well separated from their surroundings and are yellow to orange in colour. They have rich vascularization, but the vessels are often compressed by bloated adipocytes [1].
Signs and symptoms
Most lipomas enlarge slowly, and their presence does not cause any symptoms. The complications caused by lipomas are due to the mass effect and their pressure on surrounding structures. For instance, intracranial lipomas may cause diplopia, hydrocephalus, seizures, hearing impairment, and vertigo [2]. A lipoma compressing the superior vena cava can lead to oedema of the head, neck, and upper limb, as well as pain and numbness [3]. Additionally, chest lipomas can be responsible for shortness of breath, coughing, haemoptysis, and even angina and heart failure [4, 5]. In other reported cases, gastric lipomas have caused pain, nausea, vomiting, and gastrointestinal bleeding [6].
Diagnosis and treatment
The physician evaluates subcutaneous lipomas by palpation; lesions are usually non-painful, round, and mobile, with a characteristic soft, doughy feel [7]. For deep-seated lipomas, computed tomography (CT) and magnetic resonance imaging (MRI) are the best methods for visualization [2]. Sometimes other imaging studies such as radiography [8], ultrasonography [9] or much less commonly FDG-PET scan [10] and echocardiography [5] lead to the diagnosis. The pathomorphological examination uses material obtained from a fine-needle biopsy [7, 11] or a tumour resected during surgery [12]. The differential diagnosis should consider (Figures 1 B, C): liposarcoma, metastatic disease, hematoma, epidermoid cyst, nodular fasciitis, nodular subcutaneous fat necrosis, erythema nodosum, vasculitic nodules, rheumatic nodules, sarcoidosis, infections, and Weber-Christian panniculitis.
Treatment of lipomas consists in removing the lesion. Since most lipomas do not give any symptoms and the reason for treatment is aesthetic, methods that minimize the size of scars after surgery play an important role. Subcutaneous lipomas are surgically excised through an incision made in the skin overlying the mass [7]. To improve cosmetic results, surgeons use a narrow hole extrusion technique (NHET), which continues to be refined by subsequent researchers [13, 14]. Another technique used for surgical treatment is liposuction, which seems to be a good option, especially for large lipomas that would require a very long incision for classic lesion removal [15]. However, for smaller lipomas, laser lipolysis may be better [16, 17]. If the patient would like to avoid surgical treatment, injections of sodium deoxycholate [18] or corticosteroids [19] can reduce the size of lipomas. For deep-seated lipomas, the type of surgery depends on the location of the lesion and the symptoms it causes.
Genetics of sporadic lipomas
50–80% of lipomas have cells with an abnormal karyotype [20–22]. The following structural rearrangements of chromosomes, which may occur singly or in combination, have been associated with lipoma occurrence [23]: 12q13-15 region, 6p21-23 region, 13q portion loss, other loci anomalies or normal karyotype.
We most frequently observe the rearrangement of chromosome 12 in bands q13-15 [21, 22], which involve the high mobility group (HMG) protein gene located at 12q15 – HMGA2. This gene participates in the organization of the transcription complex and the regulation of transcription [24]. The translocation produces chimeric mRNA, in lipomas, this is usually HMGA2/LPP (lipoma preferred partner) [25]. The 12q13-15 aberration also occurs in other solid tumours, i.e. chondroid hamartomas [26], salivary gland adenomas, uterine leiomyomas, and endometrial polyps [27, 28]. Karyotypic alterations in chromosome 6 most often occur in bands p21-23 involving HMGA1 [27]. The protein product of this gene belongs to the same family as HMGA2 and is also responsible for regulating chromatin structure [24]. Both of these genes are abundantly transcribed in embryonic and undifferentiated cells, whereas in normal mature cells, HMGA2 should not be present at all [29, 30] and HMGA1 only in small amounts [31].
Cytogenetics as a diagnostic tool
HMGA2 expression in adipose cells allows normal adipose tissue (in which HMGA2 is absent) to be distinguished from lipoma and liposarcoma (HMGA2 present) [32]. For differentiating benign from malignant lesions, when a certain diagnosis cannot be made based on clinical and histological data, assessment of Murine double minute 2 (MDM2) gene overexpression/amplification is commonly used [33–35]. Therefore, HMGA2 staining helps pathologists make the correct diagnosis and thus select the appropriate treatment for the patient [36].
Cytogenetic changes and the clinical picture
Sreekantaiah et al. [21] analysed the cytogenetic profiles of 109 lipomas and found several associations between cytogenetic patterns and clinical parameters. Abnormal karyotypes were seen more often in older patients, in large and deep-seated lipomas, and tumours of the head, neck, and shoulders. By contrast, the results obtained by the CHAMP Study Group showed an association only with age [22]. As a potential reason for these differences, the authors suggested a different percentage of lipomas in each location compared to Sreekantaiah’s study, but they still concluded that cytogenetic changes in lipomas correlate poorly with clinical data.
Genetic diseases with lipomas
Lipomas are most often sporadic, but they can also accompany diseases caused by a germinal mutation.
Lipomas in autosomal dominant disorder
Familial multiple lipomatosis (FML) is a condition in which patients develop multiple lipomas. Most often lipomas appear in the third decade of life and their number increases until the fifth decade; numerous lesions develop gradually on the extremities and trunk [37–39]. A clinical diagnosis is made based on an evaluation of the family pedigree and the presenting symptoms of the disease in its members [37, 40]. The gene responsible for the disease has not been identified, but analysis of the pedigrees of affected families indicates autosomal dominant inheritance [40]. Unlike patients with Dercum’s disease, patients with FML do not experience pain associated with lipomas, and the most common reason for treatment is aesthetic. Treatment consists of surgical excision of the lesion using techniques that minimize scarring, liposuction, and lipolysis with phosphatidylcholine [41–43]. The differential diagnosis for FML should include other diseases in which multiple lipomas appear in the clinical picture (e.g., multiple endocrine neoplasia type 1, neurofibromatosis type 1, Dercum’s disease) and multiple symmetrical lipomatosis (Madelung’s disease). In Madelung’s disease fat tissue accumulates symmetrically on the neck, back, and upper trunk, resulting in the characteristic “horse collar” rounded appearance. It primarily affects middle-aged men who consume alcohol [44].
Another disease with autosomal dominant inheritance accompanied by lipomas is multiple endocrine neoplasia type 1 (MEN 1) – a genetic disease predisposing to parathyroid, pancreatic, and pituitary tumours. We can observe both multiple and solitary lipomas as one of the skin lesions found in these patients [45, 46]. Scientists proposed including skin lesions in the diagnostic criteria, which could facilitate the diagnosis in some complicated cases (e.g., the presence of endocrinopathies not associated with the syndrome, negative family history, poorly expressed hyperparathyroidism, unavailability of genetic tests) [45, 47]. Although lipomas are much more common in MEN 1 patients than in the general population and show high specificity, due to their low sensitivity, they cannot be one of the diagnostic criteria [45], unlike angiofibromas and collagenomas. The presence of lipomas, both subcutaneous and visceral, is found in 17% to 34% of patients [45–47]. Dreijerink et al. discussed the molecular basis of lipomas development in patients with MEN 1. They found that menin, absent in MEN1 patients, is an important factor in PPARγ (peroxisome proliferator-activated receptor)-mediated adipogenesis and that loss of PPARγ function contributes to lipoma development [48].
Lipomas also occur more frequently in neurofibromatosis type 1 (NF1) patients than in the general population as one of many skin lesions. In Miraglia’s study involving the evaluation of cutaneous manifestations of NF1 in 1102 patients, lipomas were the fourth most common skin lesion (6.2%) [49]. Typically, lipomas appear singly, but multiple cases have also been observed [49, 50].
Given that cutaneous lipomas are rare in children, their presence should prompt a complete physical examination to look for other disease manifestations of various genetic syndromes (Table 1). One of them is Bannayan-Riley-Ruvalcaba syndrome, which belongs to a group of diseases called the PTEN hamartoma tumor syndrome (PHTS) group. This autosomal dominant disease is caused by a rare developmental defect during embryogenesis which leads to macrocephaly, neurological abnormalities, hamartomatous intestinal polyposis, genital lentiginosis, lipomas, and other cutaneous lesions [51, 52]. Martin et al. highlight the diagnostic problems and suggest including the presence of lipomas among the main diagnostic criteria in children [52]. In turn, early diagnosis is important because of the increased rate of malignancy in patients [53].
Table 1
Rare genetic syndromes with lipomas in clinical presentation
Lipomas in autosomal recessive disorder
This group includes Wilson’s disease. Schaefer et al. detected subcutaneous lipomas in 21 of the 80 patients examined, with 16 cases involving multiple subcutaneous lipomas. The higher prevalence of lipomas did not associate with the severity of the disease, its presentation, or anti-copper therapy. They suggested that subcutaneous lipomas may be considered a clinical sign that could facilitate the diagnosis of this disease in some cases [54].
Mitochondrial diseases
Lipomas also occur in certain mitochondrial diseases with specific mutations. Lipomas are reported in patients with mitochondrial diabetes associated with a tRNALeu(UUR) [55] and 3243 tRNALeu(UUR) mutation [56]. Then Larsson et al. described the association between the A83446 mutation of mitochondrial DNA, MERRF syndrome and multiple symmetric lipomas [57]. Furthermore, Finsterer et al., after summarising studies by other authors, concluded that lipomas, especially cervical, are an important clinical feature found in MERRF cases [58].
Disorders of ambiguous aetiology
A disease of unclear pathophysiology that presents both sporadically and familial is Dercum’s disease (adiposis dolorosa). On the one hand, researchers suspect that an inflammatory process underlies the disease, but the findings do not provide a clear answer. Hansson et al. compared the inflammatory response in fat biopsies from Dercum’s disease patients with obese pain-free control patients and found no significant difference [59]. However, based on their research which compared blood samples, Lemaitre et al. suggested low levels of NK lymphocytes as the primary mechanism of disease development, leading to basophil activation and subsequent subclinical inflammation and pain [60]. On the other hand, the literature describes cases of familial occurrence of this disease, which indicates an autosomal dominant inheritance with different expressions [61, 62].
The characteristic of Dercum’s disease is chronic pain (>3 months) in the adipose tissue and generalized overweight or obesity. After excluding all other causes of the patient’s complaints, these features form the basis for the diagnosis [63]. Although the relationship between Dercum’s disease, chronic pain, depression, and obesity is complex, patients with adiposis dolorosa are significantly more likely to suffer from depression than obese healthy controls [64, 65]. Patients also complain of sleep disturbances, impaired memory, difficulty concentrating, anxiety, rapid heartbeat, shortness of breath, diabetes, bloating, constipation, fatigue, weakness, and joint aches [63–65].
This rare disease affects women more often than men and appears between the ages of 35 and 50 [60, 64, 66]. Cases of Dercum’s disease fall into one of four groups [63]: patients present with a generalised diffuse form (widespread painful adipose tissue without lipoma) [67], a generalised nodular form (general pain in adipose tissue and increased sensitivity surrounding lipomas), a localised nodular form (pain in and around multiple lipomas), and a juxta-articular form (painful adipose tissue near large joints) [68]. In the nodular form of the disease, multiple subcutaneous lipomas appear on the back, thighs, and arms [60, 66], which usually present histologically as ordinary lipomas [59, 61], but sometimes individual lesions can demonstrate angiolipoma or spindle cell lipoma [66].
There are no clear treatment guidelines, but the key point is pain relief. Treatment includes surgical procedures such as excision of lipomas or liposuction, as well as various pharmacological agents [63]. Excision of the lesions is often ineffective and results in only a partial improvement due to the appearance of new lipomas [69], while the long-term efficacy of liposuction is difficult to assess because of the simultaneous use of different forms of treatment by patients [67]. Pharmacological treatment includes non-steroidal anti-inflammatory drugs (which are not very effective), narcotic drugs, lidocaine (both systemic and in the form of a patch), interferon alpha-2b, corticosteroids, and calcium channel modulators, such as pregabalin and oxcarbazepine [63].
Conclusions
Genetic knowledge and testing are useful in clinical practice for lipomas. They help differentiate between normal fat tissue, lipoma, and liposarcoma. Moreover, the presence of lipomas can lead to the correct diagnosis of various diseases and, therefore, the application of appropriate treatment.