






Introduction
Ewing sarcoma (ES) is a rare, highly aggressive disease, the second most frequent bone tumor in children and adolescents, and third in the overall population [1]. Disseminated disease is detected in about 20–30% of patients at the time of initial diagnosis. Moreover, approximately 30–40% of patients develop local or distant recurrence after curative treatment [2–4]. The most frequent sites of metastases are lungs, bones, and bone marrow [5–7].
The implementation of chemotherapy in the 1960s into the curative treatment resulted in a remarkable improvement in 5-year overall survival (OS) rates in localized disease from 10% to 70–80% [7–9]. Despite this, patients with metastatic and recurrent disease have a poor prognosis, and 5-year OS rates in this group are less than 30%. Early recurrence, below 24 months from completion of curative treatment, is associated with a much worse prognosis [4].
Currently, there are no standards for the treatment of recurrent/disseminated ES. The chemotherapy of second and subsequent lines is based on the standards of the treatment of localized disease. In addition, the combination of alkylates with topoisomerase inhibitors, or irinotecan with temozolomide, or gemcitabine with docetaxel, or high-dose ifosfamide/carboplatin with etoposide are other therapeutic options; however, the duration of response is short, and the outcomes are disappointing [1, 9, 10].
New therapeutic approaches are needed to improve patient survival. Molecular target therapies are a very promising but challenging option, mainly because ES is characterized by a low mutation burden [11–13]. This is due to the oncogenic driving translocation between the EWSR1 gene and ETS family transcription factors that dominated the ES genetic landscape [11]. The most common fusion is EWSR1-FLI, observed in 80–85%, followed by EWSR1-ERG fusion [11, 14–16]. Besides characteristic fusion, additional pathogenic point mutations are observed in numerous genes, with the most common loss-of-function mutations in STAG2, TP53, and CDKN2A genes. Considering the extensively growing field of precision oncology, pathogenic variants observed in patients with ES could be potential therapeutic targets [4, 12, 17, 18]. The role of targeted therapies in ES is limited, primarily due to the sparse evidence about its mutational landscape and high heterogeneity. Thus, every effort should be made to identify genetic targets for developing novel therapies in the future [11, 18].
Our analysis aimed to assess the mutational landscape of ES in pretreatment samples, tumor samples after neoadjuvant chemotherapy, and in metastatic/recurrent tumors in children and adults. We also aimed to assess whether the mutational profile correlates with pathologic responses to neoadjuvant chemotherapy.
Material and methods
Patients
The material consisted of formalin-fixed, paraffin-embedded (FFPE) paraffin blocks of primary tumor biopsy, a surgical specimen from primary tumor resection and/or a recurrent/metastatic tumor from 5 pediatric (< 18 years old) and 17 adult patients. There were 13 (60%) males and 9 (40%) females. The median age was 24.5 years (range 7–61). Tumors were located in bones in 18 (82%) patients and soft tissues in 4 (18%) patients. All patients received neoadjuvant chemotherapy and underwent surgical resection of the tumor. Patients were diagnosed and treated in the Maria Sklodowska-Curie National Research Institute of Oncology in Warsaw (adult patients) and the Mother and Child Institute in Warsaw (pediatric patients) between 2003 and 2018.
All 22 patients had a tumor biopsy sample analyzed. Specimens from primary tumor resection after neoadjuvant chemotherapy were included for 8 of them (5 adults, 3 three children); the remaining 14 patients did not have available samples due to the lack of viable tumor cells in the specimen (caused by the response to chemotherapy). Moreover, samples from local recurrence (2 adult patients) and distant metastases (4 children, 3 adults) were included in the analysis.
For functional analysis, patients with no more than 5% of viable cells in the surgical specimen after neoadjuvant chemotherapy were classified as “responders,” while patients with more viable tumor tissue were “nonresponders”. Eight patients were classified as responders (only adult patients), and 14 were nonresponders to chemotherapy (5 children and 9 adults).
Nucleic acid isolation
Formalin-fixed, paraffin-embedded tissue sampleswe re macrodissected to obtain 100% tumor cells for nucleic acid isolation. DNA and RNA were isolated from tumor cells’ FFPE samples using the MagMax FFPE DNA/RNA Ultra Kit (Thermo Fisher Scientific, Waltham, MA, US) according to the manufacturer’s protocol. DNA and RNA quality and quantity were assessed using a NanoDrop spectrophotometer and Qubit fluorometer. DNA was stored at –20°C and RNA at –70°C.
Library preparation and sequencing
Before construction of libraries reverse transcription was conducted to obtain cDNA from RNA. DNA and cDNA libraries were prepared using Oncomine Comprehensive Assay v3 (Thermo Fisher Scientific, Waltham, MA, US) according to the manufacturer’s protocol. Oncomine Comprehensive Assay v3 is a targeted assay that enables the detection of relevant single nucleotide variants, copy number variants, gene fusions, and indels from 161 unique genes. Libraries’ concentrations were analyzed using an Agilent 2100 Bioanalyzer. Sequencing was performed using the Ion Proton instrument with the Ion PI Hi-Q Sequencing 200 Kit (Thermo Fisher Scientific, Waltham, MA, US).
Data analysis
Variants were named using Ion Reporter (version 5.10), using the “Oncomine Comprehensive v3-w3.2 – DNA and Fusions – Single Sample” protocol. Variant calling parameters were as follows: Fusion panel: “v3 Fusions v1.2”, target regions: “v3 Regions v1.1”, hotspots: “v3 Hotspots v1.1”, annotations: “v3 Annotations v1.2”, reference genome: hg19. Variant annotation was performed using CRAVAT (version 4.3) [19]. Annotated variants were loaded into R (version 3.4.1), where additional filters were applied to extract pathogenic variants: variants present in more than 2% of the population according to the Exome Aggregation Consortium database (ExAC, > 60000 exomes) [20] were removed; a variant is pathogenic according to either CHASM [21] or VEST algorithms [22] (FDR < 0.15) or the Oncomine database. All rare variants in driver genes were also considered pathogenic. Oncomine database “waterfall” plots were prepared and tumor mutation burden (TMB) was calculated using the GenVisR library (version 1.6.3) [23]. GenVisR uses the following formula for TMB estimation: mutations in sample/coverage space 1,000,000. Tumor mutation burden calculation assumed that the coverage breadth within the cohort was approximately equal. Pathogenic variant extraction was performed after TMB calculation.
The indicators used for the functional characterization of the mutation profile are calculated as follows: The percentage of mutation in a given gene in all samples was used to characterize ES in general. For comparison with the response to treatment, the proportion of mutation in the test group was subtracted from the proportion in the control group (responding to treatment) and then multiplied by the negative decimal logarithm of the p-value in Fisher’s test. Data for adults and children were analyzed in the same way – the proportion of mutation in the adults was subtracted from the proportion in children. Both ratios were calculated for genes in which the p-value in Fisher’s test for a given comparison was different from 1. To compare mutations from the biopsy and primary tumor after chemotherapy, the ratio of the number of mutations in the primary tumor to the biopsy was calculated (comparing pairs of samples). The obtained ratio was logarithmically transformed. The calculated ratios were used for functional analysis, performed using GSEA software on Hallmark and Reactome datasets. Only gene sets greater than five were taken into account. Results with a false discovery rate (FDR) < 25% and, if there were no type I errors, with a p-value < 0.05 were considered significant.
Results
The mean coverage per sample was 2290, and the median for percent of bases with the coverage more than 100x was 96%. 2505 somatic mutations were identified in 133 out of 161 genes tested in Oncomine Comprehensive Assay v3. The average number of variants per sample was 64.2. There were no significant differences in the TMB and number of variants between biopsy samples, primary tumors, local recurrences, and metastatic lesions.
For the best genomic characterization of ES, treatment-naïve biopsy samples were selected for further analysis. 1119 potentially pathogenic variants were identified in biopsy samples, resulting in an average of 50.9 mutations per patient. All patients had TMB < 10 mut/Mb, with the highest TMB of 6 mut/Mb in one patient (Fig. 1). The most commonly mutated genes were PIK3R1 (13 patients, 22 variants) and POLE (11 patients, 46 variants) (Fig. 1, Table 1). A WHSC1L1-FGFR1 gene fusion was identified in one patient. Moreover, G388R mutation of the FGFR4 gene was identified in 18 of 22 patients (81.8%), but due to high incidence in the population (30.0% in ExAC database), it is not considered pathogenic and not included in the analysis.
Fig. 1
Waterfall plot with the genes in which mutations were identified in at least 20% of biopsy samples
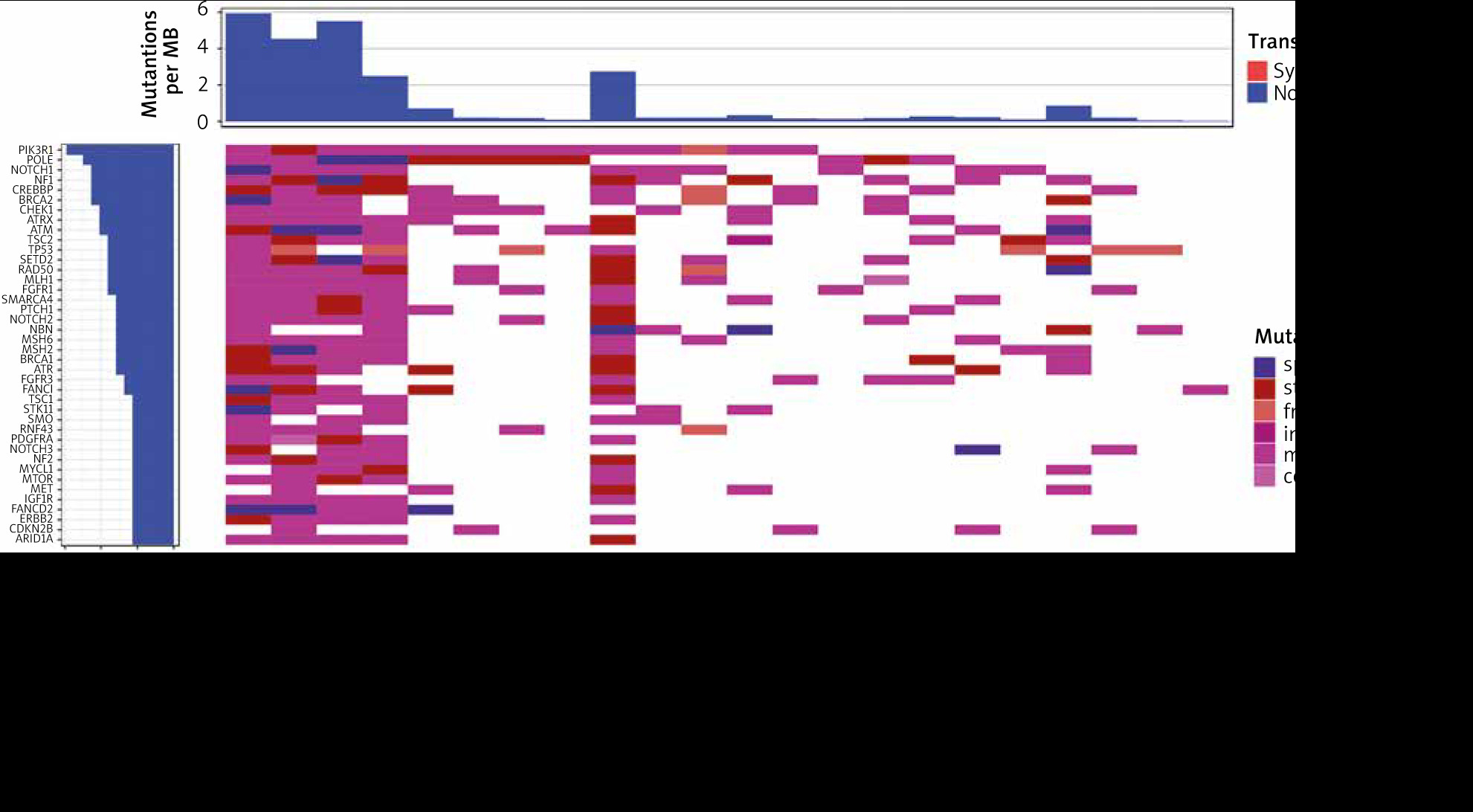
Table 1
Most commonly mutated genes and number of pathogenic variants in biopsy samples (N = 22)
The most commonly recurrent variant observed in biopsy samples were PIK3R1 T369I observed in 50% of cases, FGFR1 E159K, and TP53 at codon_72 (_72_)_, both in 27.3%. The same variants were also the most common if all sample types were included, in 63.6%, 45.5%, and 45.5% of patients, respectively (Table 2).
Table 2
Most recurrent pathogenic variants observed in at least 10% of biopsy samples
ES samples were characterized by a higher rate of mutations in the genes from four Reactome pathways and four pathways from the Hallmark collection (Table 3), with the TP53 gene being represented recurrently. Additionally, the ATR,BRCA1,RAD50,ATM,CHEK1, and NBN genes showed a significantly higher number of mutations.
Table 3
Functional annotations with over-representation of mutations in all Ewing samples, together with the level of significance and the genes that significantly affect the enrichment statistics calculated by the GSEA program (Reactome or Hallmark)
To assess whether mutational profiles predict response to chemotherapy, we compared biopsy samples from responder and nonresponder groups. Mutations in the CD28 co-stimulation and Cell cycle progression: G2/M checkpoint pathways were overrepresented in responders, while in nonresponders, mutations in the Vesicle-mediated transport pathway were overrepresented (Table 4). In comparing primary biopsy and surgical samples after neoadjuvant chemotherapy, the most mutations appearing only in the surgical specimens were in the AKT1,FLT3,PIK3CB,POLE,SETD2, and TP53 genes. Among the mutations occurring only in biopsies, the AKT3 gene had the highest absolute score value. The functional analyses revealed that mutations are overrepresented in three pathways for the surgical samples (Table 4).
Table 4
Functional annotations with over-representation of mutations in patients responding or not responding to treatment, children or adults, and biopsy or surgical specimen, together with the level of significance, and the genes that significantly affect the enrichment statistics calculated by the GSEA program (Reactome or Hallmark)
Comparing ES samples from adults and children, we found that mutations in the UV response: upregulated genes pathway were over-represented in adults, while in children in the pathways responsible for Cell Cycle, DNA Repair and Transcriptional Regulation by TP53 (Table 4).
Discussion
A fusion protein originating from gene fusions of EWSR1 genes with its various fusion partners is commonly considered a driver of ES. Additional genetic alterations, such as point mutations or copy number alterations, are widely described, but their clinical and biological value is unproven. Here we report our preliminary data of the first sequencing project of ES conducted by the Polish Sarcoma Group. Despite a low number of tested samples, we confirmed that ES is a heterogeneous disease with a broad spectrum of genetic alterations.
A low tumor mutational burden (TMB) is a general feature of sarcomas and other pediatric cancers. In our cohort, all patients had low TMB (< 10 mut/Mb), consistently with previous studies [14, 18, 24]. In other studies, relapsed tumors showed a 2- to 3-fold increased number of mutations [25]; however, probably due to a low number of relapsed/metastatic cases, we did not confirm this observation. The most commonly mutated gene profile was also consistent with available literature [18, 26–28]. Interestingly, we observed a high proportion of patients with mutated PIK3R1, NOTCH1, or CREBBP genes, suggesting their importance in ES. The gene panel used in this study did not include the STAG2 gene frequently altered in ES [29, 30], which is an important limitation of our analysis.
PIK3R1 mutations were the most frequent and were observed in over 50% of patients, mainly in adults and patients with good responses to chemotherapy. The most frequent variant in PIK3R1 was T369I, observed in most mutated cases. Its clinical importance and possible pathogenic potential have not yet been investigated. Generally, the PIK3R1 gene provides instructions for making a subunit of phosphatidylinositol 3-kinase (PI3K), an essential part of the PI3K/AKT/mTOR pathway. Multiple PIK3R1 mutations have been described as critical pathogenic drivers in other tumors, e.g., endometrial cancer. Moreover, the PI3K signaling pathway has been shown to play a particular role in phenotypes relevant to the aggressive biological behavior of ES [29], which makes this pathway a prospective therapeutic target [30]. The first clinical trials with PI3K inhibitors (NCT03458728, NCT04690725) in ES are ongoing and preliminary data are awaited.
Other genes from the PI3K/AKT/mTOR pathway, mainly AKT1,AKT3,TSC1, or TSC2, are frequently mutated in ES. We observed a higher frequency of such mutations in patients with poor responses to neoadjuvant chemotherapy. This observation suggests the potential involvement of this pathway in drug resistance. Available mTOR inhibitors could potentially be used in the treatment of ES, possibly also in the neoadjuvant setting, to increase the efficacy of chemotherapy and reduce drug resistance [31].
We found mutations in fibroblast growth factor (FGF) pathways (mainly FGFR1 and FGFR3 genes) in nearly half of the patients. None of the variants (FGFR1 E159K, FGFR3 F386L) have previously been confirmed as pathogenic, but considering the recurrent pattern of their presence in ES, their importance should be verified. Importantly, the FGF pathway has been implicated in the pathogenesis of different sarcoma subtypes, including ES. Girnita et al. suggested that it might be essential to maintain a malignant phenotype of ES cells through upregulating the EWS-FLI-1 protein [32].
We also identified a novel WHSC1L1-FGFR1 gene fusion in one patient, who had not previously been assessed for EWSR-translocation, but whose immunohistochemical staining profile was undoubtfully correlated with ES diagnosis. This genomic alteration has not previously been described in ES but has been detected in other malignant tumors, e.g., anaplastic thyroid carcinoma and lung cancer [33, 34]. Experimental data suggest that the genes WHSC1L1 and FGFR1 are responsible for the pathogenic role of the fusion protein in lung cancer and are potential targets for inhibition [33, 35]. Various FGFR inhibitors are currently being tested in clinical trials, including multikinase inhibitors covering FGFR (lenvatinib, ponatinib, regorafenib) and specific FGFR inhibitors (erdafitinib, pemigatinib). FGFR4 inhibition with a specific inhibitor (BLU9931) was highly successful in ES cell lines, suggesting the FGF pathway’s importance [36]. These drugs could offer some clinical benefit for patients with ES with WHSC1L1-FGFR1 fusion or mutations in FGF pathway genes.
Mutation in G388R in the FGFR4 gene is common in the population, observed in approximately 30% of people, and thus is not considered pathogenic. In our cohort, FGFR4 G388R was present in over 80% of patients, significantly more frequently than in general. This suggests that this variant might be associated with an increased risk of developing ES in individuals harboring this variant. Previous studies reported the FGFR4 G338R variant in around 50% of ES cases [24, 26]. Pathways noted to be altered in the presence of FGFR G388R include MAPK (33%), WNT (32%), NOTCH1 (20%), HRR (19%), and histone/chromatin remodeling (18%), all with a proven pathogenic role [24]. It is suggested that this variant may be involved in an alternative model of sarcoma genesis of ES, but this hypothesis requires further validation [26].
We also detected a higher number of mutations in genes from the DNA damage response pathway (BRCA1,BRCA2,CHEK1,ATM,PALB2), especially in children. Based on the data from other cancers, tumors harboring such mutations may respond to treatment with PARP inhibitors. Preclinical studies suggest that ES cells are sensitive to this class of drugs, especially in combination with chemotherapy [37]. The first clinical trial was conducted with olaparib but did not bring significant improvement; further trials in combinations are ongoing (NCT01858168) [11, 38].
Fifty percent of patients harbored variants in POLE genes. Mutations in POLE are generally associated with an ultramutated state in various tumors [39]. Mutations responsible for the mutator phenotype are usually located in the N-terminal exonuclease domain (e.g. P286R and V411L), and these were not found in our study, which partially explains the low TMB in our cohort. The recurrent truncating mutation Q2228* found in our dataset localizes in the distal part of the protein. POLE protein is 2286 amino acids long; therefore the mutated POLE would be 62 amino acids shorter and devoid of the putative CysB motif responsible for the formation of polymerase complexes. This conclusion is however speculative and requires further studies.
Our analyses confirmed that despite the low tumor mutational burden in ES, numerous likely pathogenic variants are detected in tumor suppressor or driver genes. Some of the mutations can act as a prognostic or predictive marker. Moreover, based on the genetic profile, targeted therapies could be potentially implemented into the treatment of ES. In recent years many clinical trials in this area have been conducted, many of which are still ongoing (Table 5).
Table 5
Summary of completed and ongoing trials with targeted therapies in Ewing sarcoma
The study has some limitations that have to be mentioned and taken into consideration when interpreting the findings. Sequencing was performed on archival FFPE tissue samples collected in routine clinical practice before commencing the project; thus, the genetic material quality was lower than that of fresh frozen samples, which could affect detection of variants. The study is prone to selection bias because only patients with available biopsy samples (untreated tumor) and adequate quality of isolated DNA were included. Moreover, only a proportion of patients had available material from surgical removal of the primary tumor or tumor recurrence/metastases, so all subgroup analysis results should be considered as hypotheses that require further validation.
Conclusions
Ewing sarcoma is a heterogeneous disease with various genetic alterations observed in the tumor cells. Distinct molecular profiles are observed between children and adults as well as between patients who respond to neoadjuvant chemotherapy and those who do not respond. Further efforts to better describe the mutational landscape of this tumor are required to understand the biology of the disease better and create opportunities for using targeted therapies to treat patients with advanced ES.