








INTRODUCTION
Neurodegeneration with brain iron accumulation (NBIA) is a group of syndromes in which iron accumulates in particular regions of the brain, mostly the globus pallidus. Individual disease entities are differentiated based on genetic, radiological, and clinical studies [1-3]. The most common type of NBIA is pantothenate kinase- associated neurodegeneration (PKAN, previously known as Hallervorden-Spatz disease), which is inherited in an autosomal recessive manner [1, 3-5]. It is estimated that the incidence of PKAN is about 1-9/1 000 000, while its etiology is still not fully understood [5]. The main limitation of the paper is the rarity of the disease the absence to date of large, multicenter, double-blinded studies.
MATERIAL AND METHODS
For this review we searched the PubMed database for articles that addressed PKAN that appeared between 2017 and 2024. The keywords used were “PKAN”, “Hallervorden-Spatz disease”, and “pantothenate kinase-associated neurodegeneration”. As a result, a total of 180 articles were identified. The papers were evaluated based on their titles, abstracts, and complete texts, with the primary inclusion criteria being the description of PKAN. The most important exclusion criterion was that the article addressed other NBIA diseases. Papers that were not written in the English language were also excluded. The review included 44 articles after the final evaluation.
ETIOLOGY AND PATHOPHYSIOLOGY
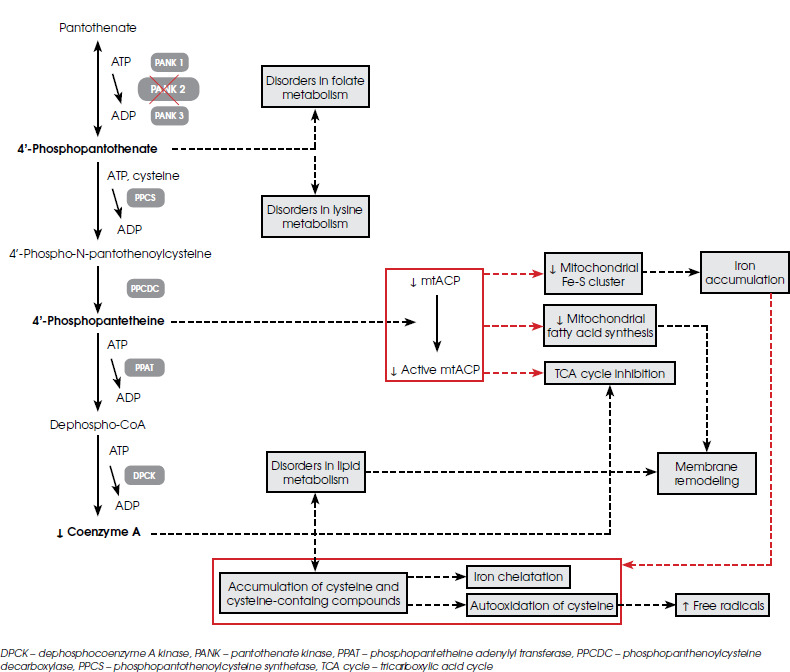
The PANK genes encode isoforms of proteins that catalyze the initial step in the biosynthesis of coenzyme A (CoA). The four mammalian PANK genes have distinct characteristics, such as tissue distribution and localization, whereas the human PANK2 gene is found in the mitochondrial intermembrane space and is abundant in the brain [1, 6, 7]. This gene is located on chromosome 20 – its exact location is 20p13 [8, 9]. A mutation in the PANK2 gene is attributed to the majority of hereditary cases of PKAN. The outcome of this mutation is a disorder of CoA metabolism, which is associated with pantothenate kinase (PANK2 enzyme) deficiency [5, 10, 11]. This enzyme uses adenosine triphosphate (ATP) to convert (R)-pantothenate into (R)-4’-phosphopantothenate, which is required for the post-translational activation of certain proteins, including the following: 10-formyltetrahydrofolate dehydrogenase, involved in folate metabolism; α-aminoadipate semialdehyde synthase in lysine metabolism; acyl carrier protein (ACP) in fatty acid synthesis (FAS) type I; and mitochondrial ACP (mtACP) in mitochondrial FAS type II [7]. A decrease in the production of CoA can result in the accumulation of cysteine and cysteine-containing compounds within the basal ganglia of the brain. This leads to the iron chelation and autooxidation of cysteine in the presence of iron, resulting in the generation of free radicals [5, 10, 11].
A significant role is played by mtACP in the production of fatty acids and the biogenesis of iron-sulfur clusters. mtACP requires 4’-phosphopantetine to be activated. The active product of this transformation is used mainly in the I complex of electron transport. It is also known that the active form of mtACP is necessary for the formation of acyl-ACP, including the lipoic acid precursor, which is involved in the activation of pyruvate dehydrogenase. Acyl-ACP is involved in the formation of iron-sulfur clusters, which are essential for the proper functioning of mitochondria and the preservation of iron equilibrium [10]. The disruption of CoA homeostasis resulting from the PANK2 mutation results in a decrease in mtACP expression [7].
Furthermore, disruption of iron homeostasis may be due to disruption of the iron-sulfur cluster and heme biosynthesis in PKAN. The function of mitochondrial enzymes is dependent on the iron-sulfur cluster, and the oxygen transport process is dependent on heme. If the efficiency of both processes is reduced, then unused iron can accumulate in the mitochondria, causing damage by generating free radicals. The disruption of the mitochondrial iron metabolism can result in cytosolic iron deficiency, which in turn leads to an increase in iron absorption, thereby enhancing the disruption of mitochondrial homeostasis [10]. It is important to note that the mechanism of increased iron uptake is unknown, and plasma levels of iron in cerebrospinal fluid, as well as ferritin, transferrin and ceruloplasmin, are normal [5].
Other metabolic effects of altering the PANK2 enzyme include mitochondrial dysfunction and low energy production, due to the disruption of such processes as the citric acid cycle, sterol and steroid biosynthesis, heme biosynthesis and beta-oxidation [2, 7, 10]. Studies have shown that patients with PKAN have decreased lipid biosynthesis and impaired bile acid metabolism. As a carrier of acetyl or fatty acid molecules, CoA is at the center of lipid-driven metabolic processes. Thus, mutations in PANK2 can result in an abnormal organization of the cell membrane [1]. In PKAN patients, red blood cells are characterized by reduced deformability and increased fragility, reduced size and a reduced phospholipid/cholesterol ratio in the cell membrane [1, 6] (Figure I).
Another hypothesis proposed to explain the accumulation of iron is the abnormal conversion of lipofuscin to neuromelanin and an insufficient activity of cysteine dioxygenase [5, 11]. The pallidum and pars reticulata of substantia nigra have a higher iron content than other parts of the brain in healthy individuals, whereas in people with PKAN the iron content in these areas is excessive [5]. Histopathological examinations indicate a rusty- brown coloration of the pallidum and pars reticulata of the substantia nigra attributed to iron deposition, as well as a diminution in the size of the caudate nuclei, substantia nigra, and tegumentums. It is also apparent that there is generalized brain atrophy [5, 12]. The microscopic characteristics of PKAN are neuroaxonal and myelin degeneration [5].
Iron plays a crucial role in the visual phototransduction cascade, especially in diseases connected with a disruption of iron homeostasis, like NBIA [13]. In NBIA, the accumulation of iron in the retina leads to the generation of harmful reactive oxygen species, resulting in retinal degeneration. Mutations in the enzyme associated with PKAN can lead to the exhaustion of coenzyme A, which is a critical molecule in cellular metabolism [13]. This depletion affects tissues with high coenzyme A consumption, such as the retina, which may contribute to the development of retinopathy in PKAN patients [13].
There are multiple motor dysfunctions connected with iron accumulation. Iron tends to accumulate in the highest concentrations in the basal ganglia, such as the mentioned globus pallidus, putamen and caudate nucleus, while some areas seems to present a lower concentration of iron [14]. Among these locations can be distinguished white matter, cortical grey matter, the and the midbrain [14]. The lowest iron levels are observed in the pons, medulla and locus coeruleus [14]. For example, the most common ataxia – Friedreich’s ataxia – is linked with the accumulation of this element in the dentate nucleus of the cerebellum [14-16].
CLINICAL MANIFESTATIONS
PKAN syndrome is characterized by progressive dystonia, extrapyramidal movement disorders, and difficulty with walking. The clinical manifestations of the disease vary individually from patient to patient [3, 5]. Other symptoms are shown in Table 1 [4-6, 12].
Table 1
Selected symptoms of pantothenate kinase-associated neurodegeneration
There is a common correlation between the age of the patient at onset and the rate of disease progression – a disease with an early onset predicts a faster rate of progression [4]. Periods of rapid deterioration are frequently followed by periods of relative stability [6]. The greater is the likelihood of Parkinsonism dominating the motor phenotype, the later is the age of onset [4]. It is worth mentioning that heterozygotes carrying the PANK2 mutation are not at increased risk of developing Parkinson’s disease [4].
There exist two distinct clinical types of PKAN, namely the classical and atypical. However, their phenotype is on a continuum [1, 3, 6, 10, 17]. The classic form, with predominant dystonia, is likely due to a complete loss of function of the pantothenate kinase 2 protein. Many children experience mild apraxia and a developmental delay before the symptoms of dystonia appear. Epileptic seizures are not common in classical PKAN [4]. Age of onset is the first decade or the beginning of the second decade. The onset of the disease has been reported across all age groups [4-6]. However, the atypical manifestation of PKAN is characterized by delayed extrapyramidal dysfunction and gradual progression of the disease process. The predominant symptoms are dysarthria, spasticity, and psychiatric disorders [5]. This form of PKAN is likely due to a partial loss of function of the pantothenate kinase 2 protein [4].
Retinal degeneration is often detected early in the disease during a comprehensive diagnostic assessment that includes electroretinogram and visual field tests [17]. The symptoms begin with night blindness (nyctalopia), followed by a gradual loss of peripheral vision, and in severe cases complete blindness. Initially, the fundus of the eye shows a speckled appearance, progressing later to the formation of bone-like structures, noticeable choroidal blood vessels, and a characteristic “bull’s-eye” pattern around the macula [17]. Patients who have a normal in an eye examination at diagnosis typically do not develop retinopathy later on [17].
The atypical form of PKAN is observed in approximately 25% of individuals, and it manifests itself later in life, specifically during the second and third decades [5].
Table 2 presents a comparison of the two forms of PKAN [1, 3, 6, 10, 17].
Table 2
Presentation of characteristics of clinical forms of pantothenate kinase-associated neurodegeneration
The study by Chang et al. [18] showed that patients with two null alleles in PANK2 showed a significantly earlier onset of disease and faster progression of PKAN symptoms. The authors defined a null allele as one that includes nonsense, frameshift, splicing mutations, intron mutations, and the deletion of a single or multiple exons. Two null alleles are more common in patients with early- onset disease than in patients with late-onset PKAN. However, the study by Li et al. [19] identified 8 PANK2 mutations. Most of the PANK2 mutation pairs resulted in a reduced expression of the PANK2 protein. Patients with a loss-of-function (LoF) mutation that completely disrupted PANK2 function or a missense mutation were more likely to have a more severe phenotype. The frequency of LoF biallelic mutations was higher in patients with early onset PKAN. Patients with homozygous mutations were found to experience gait disturbances earlier than patients with heterozygous mutations. Furthermore, patients with homozygous LoF mutations had an earlier age of onset and began having gait problems earlier than patients with homozygous or heterozygous missense/LoF mutations [19].
DIAGNOSTIC METHODS
The diagnosis of PKAN is based on radiological images and characteristic clinical symptoms, followed by genetic tests focused on the detection of the PANK2 gene, which are necessary to make the definitive diagnosis [20-22]. As magnetic resonance neuroimaging – the basic radiological test in PKAN – is nowadays commonly available, a reduction in the time to diagnosis of patients with early clinical manifestations can be observed [22]. The primary diagnostic test in people suspected of having PKAN is brain magnetic resonance imaging (MRI) using iron-sensitive sequences, including T2 images or susceptibility weighted imaging (SWI) [22]. It is important to note that SWI images show higher sensitivity for visualizing iron accumulation in brain tissue than conventional methods [23]. Another technique used to detect iron deposits in the midbrain is cranial sonography [8]. The “eye of the tiger” sign is typical for PKAN, but can also be described in other NBIAs, for example mitochondrial membrane protein- associated neurodegeneration (MPAN). It is a hyperintense spot in the middle of the globus pallidus, surrounded by a hypointense signal of the rest of globus pallidus; it is visible in axial planes [4, 24]. The hypointensive area is caused by the accumulation of iron in the globus pallidus [23]. Many patients with PKAN present a typical eye-of-the-tiger sign in their MRI evaluation, but in some the bright spot in the anterior part of the globus pallidus is obscured by a large accumulation of iron deposits.
A study by Roa-Sanchez et al. [25] revealed a more rapid decrease in the volume of the globus pallidus connected with the accumulation of iron. This confirms that this nucleus is an inhibitory center and it presents abnormal functioning in PKAN. In most patients, it is a challenge to specify the exact onset of the disease and the duration of the eye-of-the-tiger sign. There are also confirmed cases of PKAN without the eye-of-the-tiger sign. In the early stages of the disease, this sign also does not have to be present, but it is likely to be visible in more advanced cases [23, 26]. The typical picture of PKAN in MRI scans is a hyperintense center with gliosis and edema, which is surrounded by a hypointense area caused by iron accumulation in the globus pallidus [23]. On the other hand, atypical presentation in magnetic resonance imaging is visible as signal reduction without clearly recognizable round hyperintensivity in the globus pallidus [23]. In cases without hyperintense center, hyperintensive streaking was present in the central region of the globus pallidus [23]. Bilateral globus pallidus calcification was also observed [23].
If the brain MRI image is not typical for PKAN, genetic tests should be performed in patients with a high probability of having this disease [22]. However, genetic testing is done in every case, even if MRI images are typical [22]. The research conducted by Zeng [26] proved that low signals in SWI sequences and high quantitative susceptibility mapping (QSM) values can be linked with the reversed eye-of-the-tiger sign, which is when the SWI signal is low and the QSM values are significantly higher. It is especially favorable in patients who do not show the tiger’s eye sign in the early stages. In PKAN patients a hypointense signal in the substantia nigra is also detected [4].
The phenotypic spectrum of PKAN was broadened after the discovery of the PANK2 gene [27]. Goldberg et al. [28] radiologically examined patients with the eye-of-the-tiger sign and demonstrated that the sensitivity of detecting PANK2 enzyme mutations using this method was 68%. In cases where PKAN is suspected, it is advisable to pursue genetic testing for definitive diagnosis. A thorough examination should entail sequencing and deletion/duplication analyses of the PANK2 gene, the identified mutated gene associated, as we have already seen, with PKAN. Genetic data holds significant value for prognostic predictions, facilitating prenatal or pre-implantation genetic diagnosis in subsequent pregnancies, and identifying carriers within the familial context [22].
The state of patients with PKAN disease can be evaluated using the PKAN-Disease Rating Scale (PKAN-DRS). This scale quantifies specific factors such as dystonia, rigidity, and overall motor function to provide a comprehensive assessment of the disease’s advancement and its influence on a patient’s everyday functioning. By employing this scale, medical professionals and researchers can obtain valuable insights into the condition, facilitating more informed decisions regarding treatment and care strategies [27]. Another scale used to assess patients is the pantothenate kinase-associated neurodegeneration activities of daily living (PKAN-ADL), which aims to measure the functional capacity and daily activities of individuals suffering from PKAN. It encompasses aspects such as the ability to perform daily tasks, including speaking, eating, dressing, maintaining personal hygiene, writing, chewing, salivating, drooling, sitting, changing position in bed and walking. This scale serves to provide a detailed assessment of patients’ capabilities and the impact of the disease on their daily functioning [29].
The wide range of symptoms and frequent occurrence of atypical and non-specific clinical presentation make the differential diagnosis of PKAN based on symptoms challenging. Patients can be diagnosed using MRI T2 scans in axial projections, which can help to distinguish PKAN from other NBIA diseases, such as phospholipase A2-associated neurodegeneration, MPAN, and beta propeller protein-associated neurodegeneration [4]. These conditions are mostly distinguished based on iron accumulation locations, which are different for each disease [4]. Exact diagnosis can be made after genetic testing, because of the different gene mutations and malfunctioning in various NBIA conditions [4].
TREATMENT METHODS
Currently, there are no effective treatment methods available for individuals with PKAN and patients can only be offered treatment for their symptoms [30]. Most of the existing medications do not halt the progression of the disease.
Pharmacological treatment can be divided into two groups – disease modifying agents, such as iron chelators, and symptomatic treatment agents [31, 32]. Among the drugs used for symptom-relief are anticholinergics, botulinum toxin, clonidine, gabapentin, baclofen, pregabalin, benzodiazepines and tetrabenazine [22, 31-33].
As mentioned above, one method of treating PKAN is iron chelation [10]. Frequently used agents for this purpose include deferiprone, deferoxamine and deferasirox [22, 33]. The first substance, administered orally, has the ability to traverse both the blood-brain barrier, eliminating iron and preventing its accumulation [30, 33]. Deferiprone (DFP) seems to be well tolerated by patients and efficient in decreasing iron levels in the basal ganglia, with a slight slowing of the disease progression [8, 34]. Romano et al. [30] assessed the safety and effectiveness of DFP in doses of 15 mg/kg in 10 patients with NBIA. Their research revealed a decrease in cerebral iron accumulation, documented with magnetic resonance images, followed by stability in the clinical picture of the patients. The reduction of iron accumulation in brain tissues was measured by comparing initial and follow-up MRI scans with a mean follow-up time of 5.5 ± 2.3 years [30]. It is worth mentioning that scientists did not report significant correlation between clinical and radiological presentations [30], and also that if iron chelating medicaments are not bringing favorable results, this strategy may be combined with other management patterns [10].
Another method is the administration of vitamin B. Animal studies suggest that vitamin B5 (pantothenate) may have the potential to help patients by enhancing CoA levels and reversing mitochondrial dysfunction. This treatment has demonstrated the prevention of various neurological symptoms in PKAN animal models on a high-fat ketogenic diet [33]. Vitamin B5 is recommended in all adult patients with PKAN, regardless of their phenotype, but it is not favorable in children [22]. It is recommended to administer vitamin B5 for at least three months, with a starting dose of 250 mg taken orally, with weekly increases in the dose of 500 mg, until a dose of 2-5 g daily is obtained or until adverse effects present themselves [22].
The FORT is a randomized, double-blind, placebo- controlled clinical trial with the main aim of assessing the effectiveness and safety of the administration of fosmetpantotenate in adults and children suffering from PKAN [35, 36]. The study was conducted with 84 patients randomly divided into placebo and fosmetpantotenate groups. In the placebo group, two patients experienced progression of the disease, two failed to adhere to the regimen and two died as a result of respiratory failure and complications after feeding aspiration [36]. The general conclusion from this trial was that fosmetpantotenate was safe for the patients, but that there was a lack of improvement of their neurological functions [36].
A study conducted by Alvarez-Cordoba et al. [37] suggests that commercially available supplements, such as pantothenate, pantethine, vitamin E, omega 3, carnitine and thiamine are able to reverse the accumulation of iron, followed by an enhancement of PANK2, mtACP and NFS1 expression. The correction of mutate fibroblasts with PANK2 expressions can also be achieved as a result of treatment with alpha-lipoic acid [38]. However, further research through clinical trials is necessary to evaluate its actual impact [33]. Due to isolated reports suggesting a deterioration in the condition of patients associated with supplementation with selenium, vitamin E, and idebenone, scientists do not recommend administering these compounds to individuals with PKAN [22].
Among other methods that may be implemented when treating PKAN there is deep brain stimulation, which in most cases has replaced previously utilized surgical techniques such as ablative pallidotomy or thalamotomy [22, 33]. However, these methods are still used, most commonly in combination alongside deep brain stimulation [33]. Although the disease tends to be progressive, positive outcomes observed immediately after the surgery are not permanent [33]. PKAN can be managed with the administration of intrathecal baclofen, especially in patients with severe symptoms of dystonia or spasticity [33].
Bilateral deep brain stimulation (DBS) targeting the globus pallidus in patients with PKAN, which is favorable especially in cases with dystonia, has been recommended in the literature [39, 40]. Potential targets for DBS may also be subthalamic nuclei, from which patients with notable symptoms in the limbs can benefit the most [41]. Wilson and colleagues indicated that cannabis was also used in the treatment of children with PKAN. However, there is a lack of scientific evidence and controlled placebo clinical trials regarding the use of cannabis in this disease [42].
Symptomatic treatment in PKAN may be based on various psychotropic agents, anticholinergics or botulinum toxin [22, 31-33]. Botulinum toxin type A is effective in adults suffering with the disease, especially if they present dystonia and spasticity [32]. In the study conducted by Hameed et al. [31], physicians injected 300-units of botulinum toxin type A into the gastrocnemius muscles of patients’ legs bilaterally, with a favorable effect in reducing dystonia and spasticity. For laryngeal dystonia and respiratory issues, bilateral injections to the thyroarytenoid muscles can be given, but it is worth remembering that the full effect is delayed [22]. Other indications for botulinum toxin injection are oral dystonia and blepharospasm [22].
Management strategies for PKAN include the restoration of phosphopanthenate levels in cells, though such therapies are still at the stage of clinical studies [33]. Another method is the activation the biosynthesis of CoA by activating the PANK3 molecule, to improve abnormalities in PANK2, which are the underlying cause of PKAN [10, 33]. Gene therapy also seems to be a promising method of managing PKAN; the main limitation here is the difficulty in developing vectors capable of crossing the blood-brain barrier, but strategies based on use of adeno-associated vectors seem to be feasible [10, 33]. There is also a possibility of providing the stereotactic infusion of gene therapies for patients who have undergone a craniotomy [33].
The future of PKAN treatment appears to be precision medicine, which involves personalized treatment methods tailored to the specific patient and their symptoms, as well as customized materials based on their genetics and environmental factors [43]. Contemporary cell models indicate the potential for their use in studying the impact of drugs and their interactions on improving mutated cellular phenotypes [44]. Another promising method which may lead to a further evaluation of PKAN pathophysiology and the development of diagnostics for it is assessing iron accumulation in neurons and astrocytes [2]. α-lipolic acid also shows potential in reversing all abnormalities in mutant fibroblasts with PANK2 enzyme expression, as this substance is believed to enhance the level of PANK2 expression itself [38]. This process leads to the correction of mutant forms [38]. α-lipolic acid presents good oral bioavailability and can cross the blood-brain barrier with no significant toxic effects [38]. It is important to note that atypical presentations of PKAN disease also require an individualized approach and careful diagnostics. Further studies on iron accumulation dysfunction are also important to achieve greater knowledge of the pathogenesis of PKAN [10].
CONCLUSIONS
PKAN is a rare condition caused by iron accumulation in the brain. The precise mechanism of this disease remains incompletely understood. However, some clinical manifestations of PKAN can be explained by the biochemical effects of PANK2 enzyme deficiency, which leads to mitochondrial dysfunction, CoA deficiency and lipid metabolism disorders. Diagnosis is made on the basis of magnetic resonance images, clinical picture and genetic evaluation. MRI images commonly present a typical eye-of-the tiger sign. Management is of symptoms only, as there is as yet no effective treatment. Pharmacological methods can be divided into symptomatic medications, such as pregabalin, gabapentin or botulinum toxin, and disease modifying agents, such as iron chelators. Among other methods surgery and/or deep brain stimulation can be mentioned. The future of the management of PKAN is personalized treatment based on genetic and environmental factors.