

Introduction
Acute kidney injury (AKI) is a commonly observed organic impairment resulting from sepsis, contributing to more than 60% of mortality cases in the intensive care unit [1-3]. Recent research indicates that the inflammatory response initiated by sepsis plays a crucial role in the development of sepsis-associated AKI (SA-AKI) [4, 5]. Therefore, it is essential to conduct further investigations focused on elucidating anti-inflammatory mechanisms and developing treatment strategies targeting inflammation to improve the management of SA-AKI.
Nuclear factor κ-B (NF-κB) plays a pivotal role as a transcriptional regulator in the modulation of pro- inflammatory cytokine production and leukocyte recruitment [6]. Under normal physiological conditions, NF-κB interacts with inhibitory molecules from the IκB family and remains localized within the cytoplasm; however, in pathological states, activation of IκB kinases (IKKs) leads to ubiquitination of NF-κB, causing its translocation to the nucleus and consequently augmenting its transcriptional activity [7, 8]. Extensive research has provided evidence that the excessive activation of NF-κB in response to lipopolysaccharide (LPS) stimulation is associated with the release of pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-β, and IL-6 [9-11]. The involvement of a novel signaling cascade, specifically the NF-κB-mediated transcriptional regulation of microRNA (miR) genes, has been implicated in the induction of cell apoptosis and inflammation caused by LPS [12-14]. For example, the activation of NF-κB by LPS results in the up-regulation of multiple miRs in human biliary epithelial cells [12]. In a murine model of SA-AKI and LPS-stimulated renal tubular cells, the down-regulation of miR-376b mediated by NF-κB contributes to the inflammatory response, tubular cell apoptosis, and renal tissue damage [13]. These findings suggest that NF-κB-mediated miRs may play a significant role in the pathogenesis of sepsis-induced AKI.
Hyperforin (HYP), a prominent constituent of Hypericum perforatum, has been confirmed to possess notable pharmacological properties in ameliorating depressive disorders, cerebral nerve injuries, and inhibiting the progression of carcinogenesis [15-18]. Moreover, the anti-inflammatory potential of HYP has been demonstrated in diverse experimental models [19-22]. HYP may inhibit renal fibrosis through the PI3K/AKT/ICAM1 axis in a mouse model of unilateral ureteral obstruction, commonly utilized as an in vivo model for studying renal fibrosis [23]. This study presents evidence indicating that HYP may confer protection against renal fibrosis; however, additional research is necessary to comprehensively elucidate its effects in various in vitro and in vivo models. Consequently, the primary objective of this study is to examine the pharmacological impacts of HYP in LPS-induced inflammation and AKI, while also elucidating the molecular mechanism responsible for HYP-induced renoprotection and anti-inflammatory activity in both in vivo and in vitro septic models.
Material and methods
Animal experiment
Experiment 1. A total of 24 C57BL/6J mice (8 weeks old; body weight: 20 ±2 g) were divided into two groups (Con or HYP treated group; n = 12 in each group) to investigate the toxicology of HYP in vivo. Mice were treated with or without HYP (20 mg/kg/day; HPLC ≥ 85%; Sigma-Aldrich, Merck KGaA, Germany) for 14 days.
Experiment 2. A total of 36 C57BL/6J mice were divided into three groups: LPS group (20 mg/kg); HYP group treated with LPS (20 mg/kg) combined with HYP (20 mg/kg/day); TPCA-1 group treated with LPS (20 mg/kg) combined with TPCA-1 (20 mg/kg/day). Preventative treatment of HYP or TPCA-1 intragastric administration was implemented for 1 week before LPS injection. The survival rate was analyzed in the next four days after LPS injection with HYP or TPCA-1 treatment.
Experiment 3. A total of 48 C57BL/6J mice were divided into four groups: Con group with normal saline; LPS group (20 mg/kg); HYP group treated with LPS (20 mg/kg) combined with HYP (20 mg/kg/day); TPCA-1 group treated with LPS (20 mg/kg) combined with TPCA-1 (20 mg/kg/day). After 24 hours of LPS injection, blood, urine and kidney were collected for experimental analysis.
Experiment 4. A total of 24 C57BL/6J mice were divided into the groups: the LPS group (20 mg/kg); the antagomiR-21 group treated with LPS (20 mg/kg) combined with antagomiR-21 (20 nM/0.1 ml; twice/week; RiBo Biotech Co., Ltd., Guangzhou, China). Preventative treatment of antagomiR-21 by tail vein injection was implemented for 1 week before LPS injection. The survival rate was analyzed in the next four days after LPS injection.
Experiment 5. A total of 72 C57BL/6J mice were divided into six groups. After LPS-injected mice had been treated with normal saline, antagomiR-Con and antagomiR-21, blood and urine were collected for experimental analysis after 24 hours of LPS injection.
Histologic examination
Hematoxylin and eosin (H&E; Beyotime Biotechnology, Haimen, China) staining was performed according to standard operating procedures as described previously [24], and the tubular injury score was determined as described previously [24]. The area of injury was assessed, and semi-quantitative evaluations were conducted on renal tubule dilatation, tubule epithelial cell exfoliation, interstitial congestion, and tubular protein tube type. In brief, six tubular areas in each section were randomly selected and scored on a scale of 0 to 4 (0, < 5%; 1, 5-25%; 2, 25-50%; 3, 50-75%; 4, > 75%).
Biomarkers in serum and urine
Blood urea nitrogen (BUN), serum creatinine (Cr), urinary kidney injury molecule 1 (KIM1) and neutrophil gelatinase-associated lipocalin (NGAL) (Elabscience Biotechnology Co., Ltd., Wuhan, China) were measured according to the manufacturer’s instructions. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST; Nanjing Jiancheng Bioengineering Institute, Nanjing, China) were measured using enzyme-linked immunosorbent assays (ELISA) according to the manufacturer’s instructions. Tumor necrosis factor-α, IL-β, IL-6 (Elabscience Biotechnology Co., Ltd., Wuhan, China) were measured according to the manufacturer’s instructions.
RT-qPCR
Standard reverse transcription-quantitative polymerase chain reaction (RT-qPCR) procedures were performed as described previously [25]. The PCR primers were as follows: forward 5'-GCACCGTCAAGGCTGAGAAC-3' and reverse 5'-CAGCCCATCGACTGGTG-3' for miR-150-3p; forward 5'-CTCGCTTCGGCAGCACA-3' and reverse 5'-AACGCTTCACGAATTTGCGT-3' for U6; forward 5'-AGCAGTCGGTACAACTTAAAGG-3' and reverse 5'-ACTCGACAACAATACAGACCAC-3' for KIM1; forward 5'-TGGCCCTGAGTGTCATGTG -3' and reverse 5'-CTCTTGTAGCTCATAGATGGTGC-3' for NGAL; forward 5'-CAGGCGGTGCCTATGTCTC-3' and reverse 5'-CGATCACCCCGAAGTTCAGTAG-3' for TNF-α; forward 5'-GAAATGCCACCTTTTGACAGTG-3' and reverse 5'-TGGATGCTCTCATCAGGACAG-3' for IL-β; forward 5'-CTGCAAGAGACTTCCATCCAG-3' and reverse 5'-AGTGGTATAGACAGGTCTGTTGG-3' for IL-6; forward 5'-AGGTCGGTGTGAACGGATTTG-3' and reverse 5'-GGGGTCGTTGATGGCAACA-3' for glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Western blot
Standard immunoblotting was performed as described previously [26]. NF-κB/p65 (Cell Signaling Technology; cat. no: #59674), TNF-α (cat. no: #11948; dilution: 1:2,000; Cell Signaling Technology), IL-1β (cat. no: #31202; dilution: 1:2,000; Cell Signaling Technology) and IL-6 (cat. no: #12912; dilution: 1:1,000; Cell Signaling Technology) primary antibodies were used to incubate protein membrane. Histone (Cell Signaling Technology; cat. no: #9715) and β-actin (Abcam; cat. no: ab179467) signals were used as an internal reference. Protein bands were obtained using an ECL chemiluminescence kit (Santa Cruz Biotech, Santa Cruz, CA, USA) with the Bio-Rad Gel Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Quantitative data were analyzed using Quantity One software version 4.5 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cell culture
Mouse renal podocytes were obtained from the National Infrastructure of Cell Line Resource. Podocytes were maintained in RPMI-1640 (Invitrogen, USA) containing different concentration LPS (0, 0.1, 1, 5 ng/ml) supplemented with 10% FBS (Invitrogen, USA) at 37oC in a humidified incubator (Thermo, USA), 5% CO2, 95% air atmosphere.
Cell transfection and luciferase reporter assay
Si-NF-κB, miR-21 inhibitors, mimics, CMV-Con and CMV-p65 were synthesized by Sangon Biotech (Shanghai, China) and transfected into podocytes according to the manufacturer’s instructions. After transfection with si-Con or si-NF-κB into miR-21 transcription start site (TSS) wild-type (WT) and mutant-type (Mut) podocytes, luciferase activity was measured using a dual-luciferase reporter assay kit (Beyotime Institute of Biotechnology). In addition, in podocytes transfected with CMV-Con or CMV-p65, or stimulated with LPS (1 ng/ml), luciferase activity was measured using a dual-luciferase reporter assay kit (Beyotime Institute of Biotechnology).
Cell counting kit 8 (CCK8)
After podocytes had been treated with different experimental conditions, cell viability was evaluated by CCK8 kits (Dojindo, Japan) according to the manufacturer’s instructions. The absorbance was measured at 450 nm with a SpectraMax M5 ELISA plate reader (Molecular Devices, LLC, Sunnyvale, CA, USA).
Cell apoptosis
After podocytes had been treated with different experiment conditions, cell apoptosis was evaluated using TUNEL staining (Roche) according to the manufacturer’s protocol. TUNEL positive staining cells were counted to assess apoptotic cell proportions.
Statistical analysis
Data are presented as mean ± standard deviation. Statistical analysis was performed using GraphPad Prism Version 7.0 (GraphPad Software, Inc., La Jolla, CA, USA). Student’s t-test was used to analyze two-group differences. Inter-group differences were analyzed by one-way analysis of variance, followed by Tukey’s post hoc analysis. Survival rates were calculated using the Kaplan-Meier method with the log-rank test applied for comparison. A p value less than 0.05 indicates a significant difference.
Results
HYP toxicology in vivo
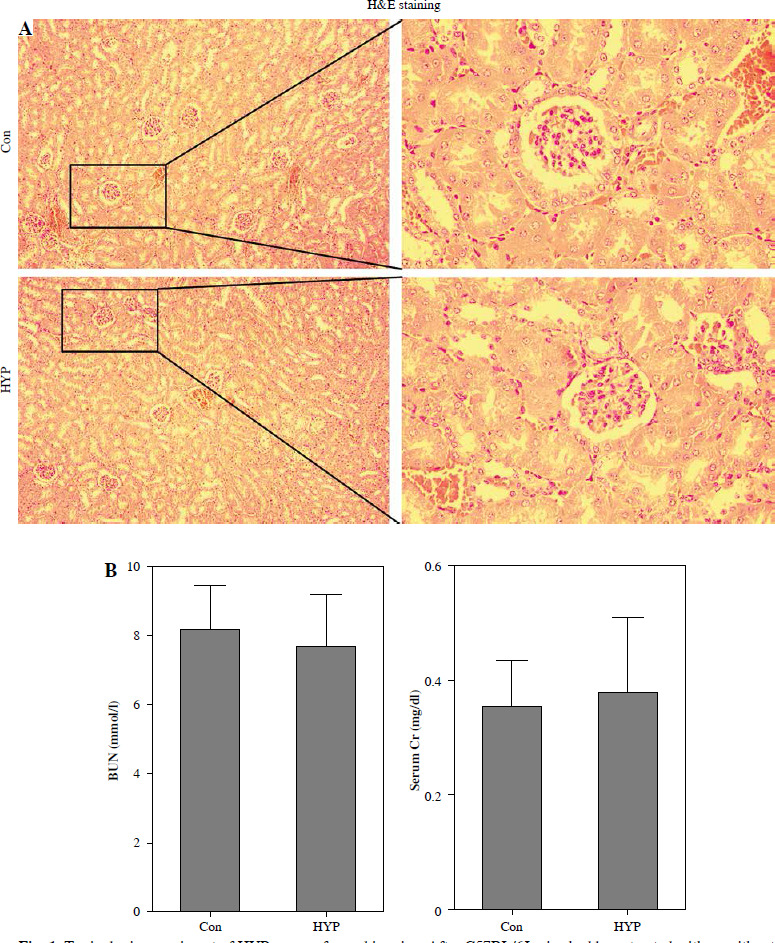
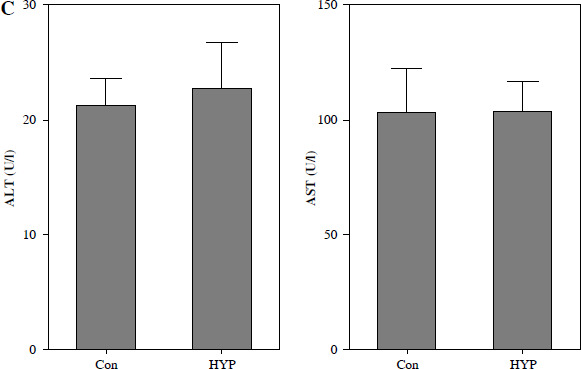
The renal toxicity of HYP was evaluated in a 14-day follow-up study. Fig. 1A illustrates the absence of histopathological changes in the HYP group compared to the control group. Additionally, the renal injury markers BUN and serum Cr (Fig. 1B), along with the hepatic injury markers ALT and AST (Fig. 1C), did not display significant deviations from their initial levels. These findings suggest that the administration of HYP did not induce any observable renal harm.
Fig. 1
Toxicologic experiment of HYP was performed in mice. After C57BL/6J mice had been treated with or without HYP intragastric administration for 14 days, H&E staining was used to perform renal histologic examination (A); renal injury biomarkers BUN and serum Cr (B) and hepatic injury biomarkers ALT and AST (C) were measured to evaluate whether HYP treatment was able to induce renal and hepatic injuryFig. 2. HYP improves survival rate and renal injury in LPS-stimulated mice. Preventative treatment with HYP (20 mg/kg/day) or TPCA-1 (20 mg/kg/day) intragastric administration for 1 week before LPS injection; survival rate was analyzed in the next four days after LPS injection (A; n = 12 in each group). After 1 day of LPS injection combined with HYP (20 mg/kg/day) or TPCA-1 (20 mg/kg/day) preventative treatment for 1 week, renal histologic examination was performed and tubular injury score was determined for the kidney of septic mice with or without HYP or TPCA-1 treatment (B)
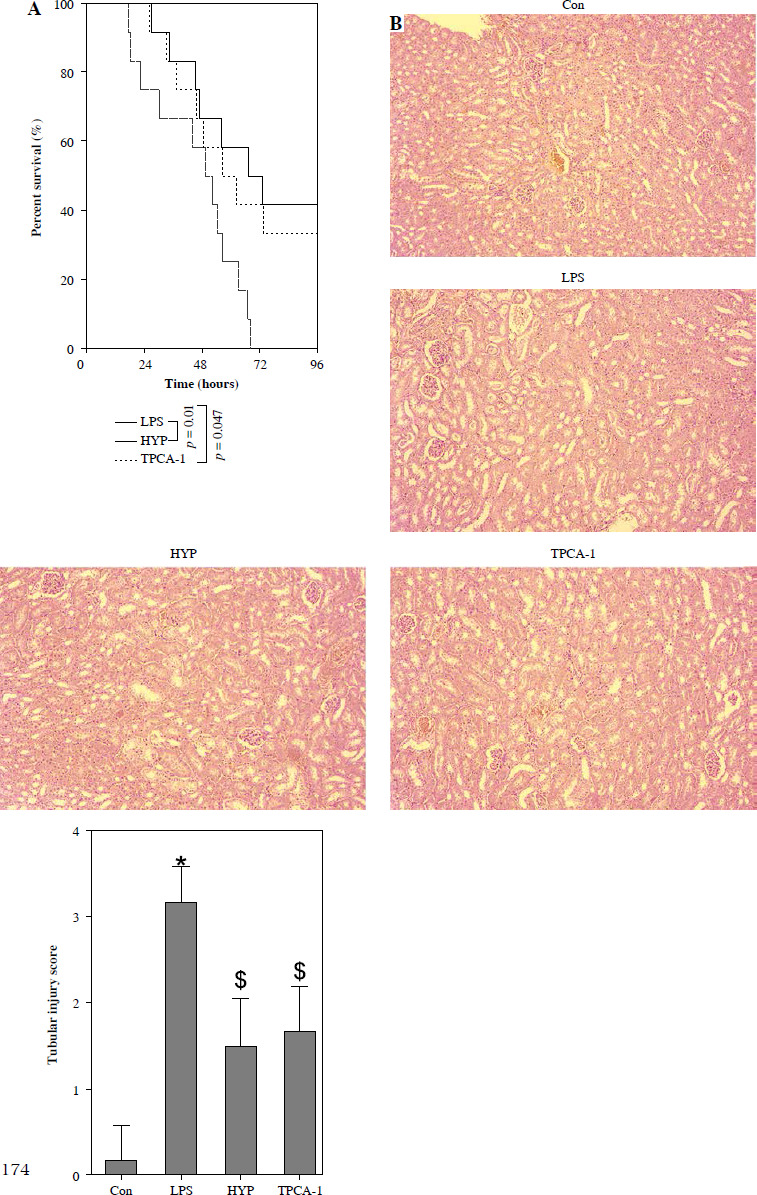
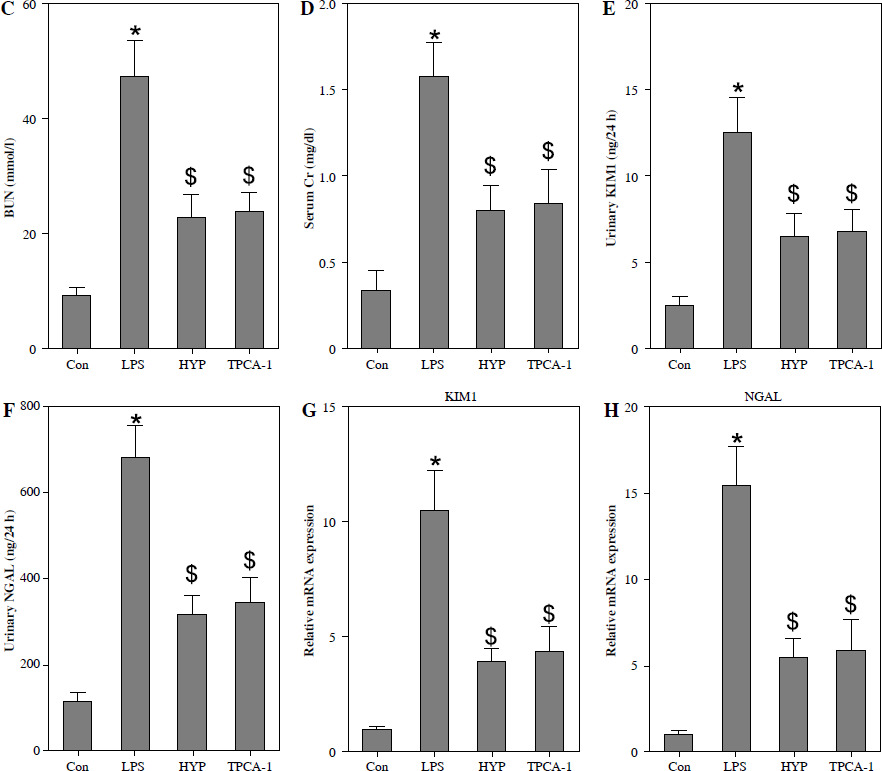
HYP improves the survival rate and renal injury in LPS-stimulated mice
To examine the potential protective impact of HYP on sepsis-induced mortality in mice, a preventive supplementation study was undertaken, wherein HYP or TPCA-1, an inhibitor of NF-κB signaling, was administered two weeks prior to LPS injection. The survival rate of mice was evaluated within a 96-hour period following LPS treatment. As illustrated in Fig. 2A, the administration of HYP or TPCA-1 resulted in significantly longer survival time and improvement in the survival rate (41.7% or 33.3%) compared to the LPS group (0%) within the designated 96-hour timeframe. The histological examination of LPS-treated mice revealed the presence of renal tubular injuries, specifically tubular atrophy and loss of epithelial cells (Fig. 2B). Furthermore, the LPS-treated mice exhibited significantly higher levels of the acute injury indicators BUN and serum Cr compared to the control group. The BUN level in the LPS-treated group was 47.20 ±5.18 mmol/l, while in the control group it was 9.27 ±1.18 mmol/l. Similarly, the serum Cr level in the LPS-treated group was 1.56 ±0.17 mg/dl, whereas in the control group it was 0.34 ±0.09 mg/dl. However, the administration of HYP or TPCA-1 resulted in a significant decrease in BUN and serum Cr levels in mice treated with LPS. The levels of HYP were 22.77 ±3.27 mmol/l and 0.80 ±0.12 mg/dl, while the levels of TPCA-1 were 23.90 ±2.65 mg/dl and 0.84 ±0.16 mg/dl, respectively (Fig. 2C, D). Additionally, the up-regulation of urinary KIM1 and NGAL (Fig. 2E, F), as well as renal KIM1 and NGAL mRNA expression (Fig. 2G, H), was significantly reduced by the administration of HYP or TPCA-1.
Fig. 2
HYP improves survival rate and renal injury in LPS-stimulated mice. Preventative treatment with HYP (20 mg/kg/day) or TPCA-1 (20 mg/kg/day) intragastric administration for 1 week before LPS injection; survival rate was analyzed in the next four days after LPS injection(A; n = 12 in each group). After 1 day of LPS injection combined with HYP (20 mg/kg/day) or TPCA-1 (20 mg/kg/day) preventative treatment for 1 week, renal histologic examination was performed and tubular injury score was determined for the kidney of septic mice with or without HYP or TPCA-1 treatment (B) Biochemical parameters BUN (C), serum Cr (D), urinary KIM1 (E) and NGAL (F) were determined to evaluate AKI. KIM1 (G) and NGAL (H) mRNA expression levels in the kidney were measured using RT-qPCR
*p < 0.05 compared with control group; $p < 0.05 compared with LPS group.
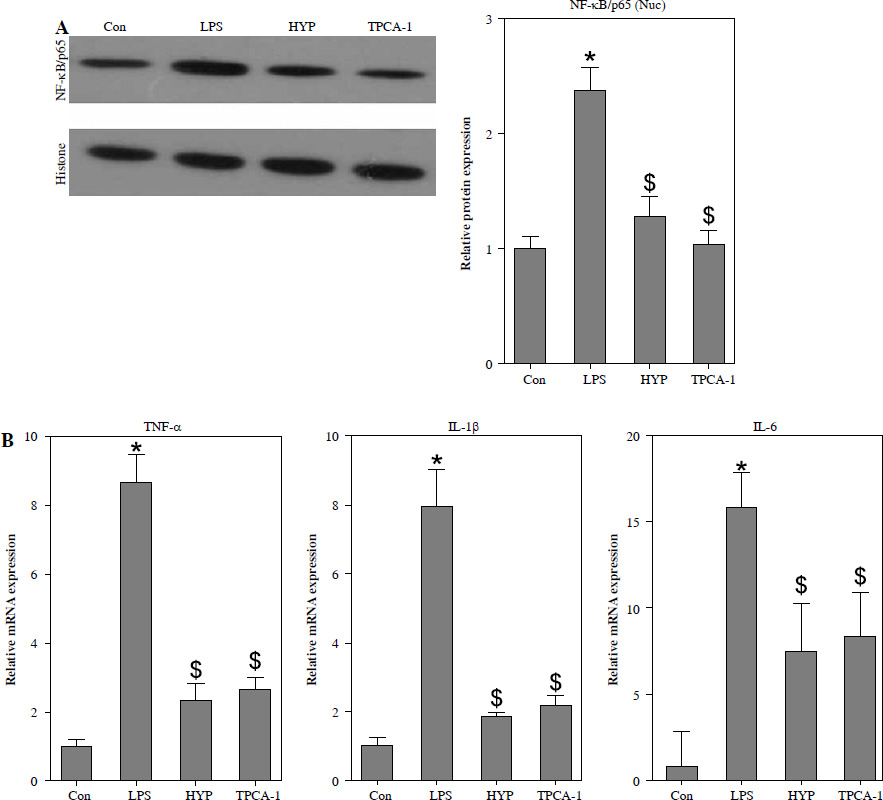
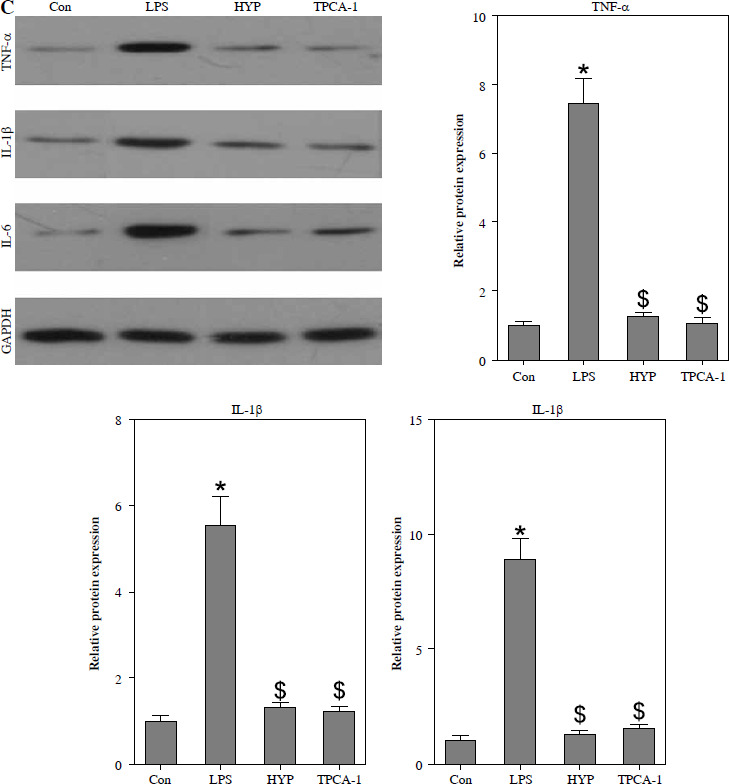
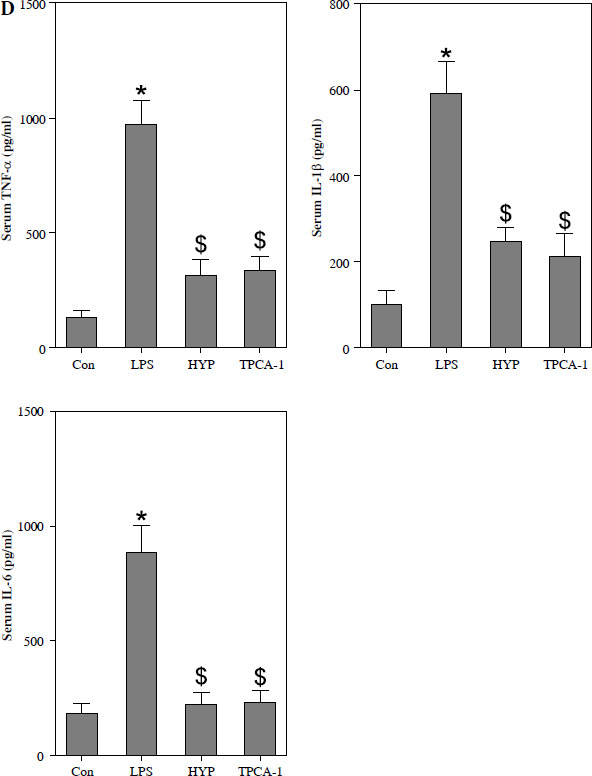
HYP inhibits the NF-κB-mediated inflammatory response in LPS-treated mice
As depicted in Fig. 3A, the injection of LPS resulted in higher nuclear NF-κB/p65 levels in the kidney compared to the control group. However, the administration of HYP or TPCA-1 significantly suppressed the expression levels of LPS-activated NF-κB/p65 in the nucleus. The findings from RT-qPCR (Fig. 3B), western blot (Fig. 3C), and ELISA (Fig. 3D) assays demonstrated that NF-κB/p65 played a role in the up-regulation of inflammatory cytokines, namely TNF-α, IL-1β, and IL-6, in the kidney or serum of LPS-treated mice. Notably, the administration of HYP or TPCA-1 attenuated the up-regulation of these inflammatory cytokines, indicating that HYP possesses anti-inflammatory activity in septic mice.
Fig. 3
HYP inhibits NF-κB-mediated inflammatory response in LPS-treated mice. Preventative treatment with HYP (20 mg/kg/day) or TPCA-1 (20 mg/kg/day) intragastric administration for 1 week before LPS injection, NF-κB/p65 protein level in the kidney was measured using western blot after 1 day of LPS injection (A); inflammatory cytokines, TNF-α, IL-1β and IL-6, mRNA (B) and protein C) in the kidney and in the serum (D) were evaluated using RT-qPCR, western blot and ELISA assays, respectively
*p < 0.05 compared with control group; $p < 0.05 compared with LPS group.
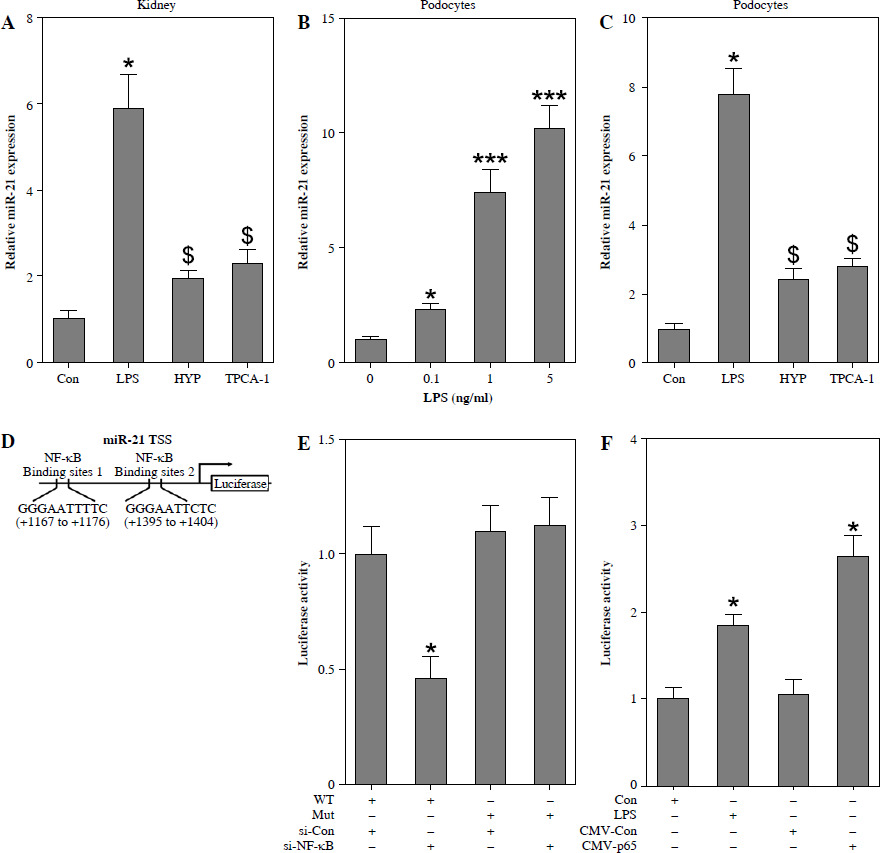
NF-κB mediates miR-21 expression in LPS-treated podocytes
A previous study demonstrated that NF-κB/p65 plays a role in facilitating the transactivation of miR-21 in human biliary epithelial cells upon stimulation with LPS [12]. In our study, we observed up-regulation of miR-21 in the kidneys of mice treated with LPS (Fig. 4A) as well as in LPS-stimulated podocytes (Fig. 4B). However, the administration of HYP or TPCA-1 resulted in significant repression of miR-21 expression in the kidneys of LPS-treated mice (Fig. 4A) and LPS-stimulated podocytes (Fig. 4C). According to the findings depicted in Fig. 4D, two binding sites existed between the miR-21 TSS and NF-κB. Subsequent luciferase assays demonstrated a significant reduction in luciferase activity in wild-type podocytes following transfection with si-NF-κB. Conversely, luciferase activity was notably increased in podocytes treated with LPS (Fig. 4E) or transfected with CMV-p65 (Fig. 4F). These results suggest that overexpression of NF-κB/p65 has the potential to enhance the transcription of miR-21.
Fig. 4
NF-κB mediates miR-21 expression in LPS-treated podocytes. Preventative treatment with HYP (20 mg/kg/day) or TPCA-1 (20 mg/kg/day) intragastric administration for 1 week before LPS injection, miR-21 level in the kidney was measured using RT-qPCR after 1 day of LPS injection (A); after podocytes’ exposure to LPS (0, 0.1, 1 and 5 ng/ml) for 24 h, miR-21 level was measured using RT-qPCR (B); after podocytes’ exposure to LPS (1 ng/ml) for 24 h combined with HYP (30 μM) or TPCA-1 (30 μM) treatment, miR-21 level was measured using RT-qPCR (C); two binding sites between miR-21 TTSS and NF-κB (D). After transfection with si-Con and si-NF-κB into WT or Mut podocytes, luciferase assay was performed to evaluate the association between miR-21 and NF-κB (E). After transfection with CMV-Con or CMV-p65 into podocytes, luciferase assay was performed to evaluate the association between miR-21 and NF-κB (F)
*p < 0.05; ***p < 0.001 compared with control group; $p < 0.05 compared with LPS group.
Knockdown of NF-κB inhibits LPS-induced podocyte apoptosis
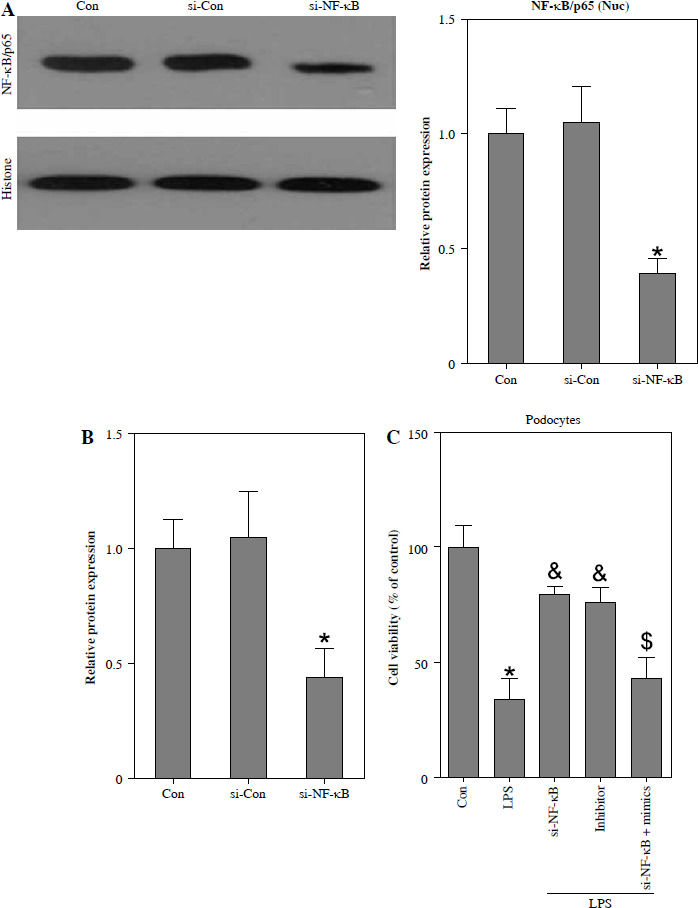
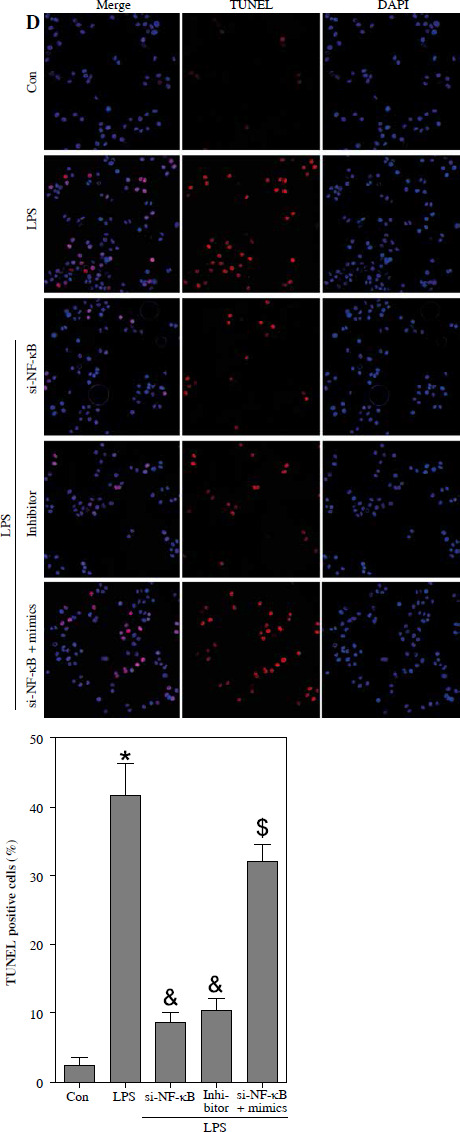
To investigate the potential association between NF-κB and LPS-induced podocyte death, transfection of si-NF-κB was performed on podocytes. The results depicted in Fig. 5A and B demonstrate a significant reduction in NF-κB/p65 protein levels within the nucleus, as well as a decrease in miR-21 expression, following si-NF-κB transfection in podocytes. Furthermore, Fig. 5C illustrates notable inhibition of cell viability upon LPS stimulation. However, the inhibition of podocyte growth induced by LPS was effectively reversed through the transfection of si-NF-κB or miR-21 inhibitor. Additionally, the protective effect of si-NF-κB in LPS-treated podocytes was counteracted by the transfection of miR-21 mimics. The induction of podocyte apoptosis by LPS was also counteracted through the transfection of si-NF-κB or miR-21 inhibitor. Nevertheless, the transfection of miR-21 mimics nullified the protective effect of si-NF-κB against apoptosis in podocytes treated with LPS (Fig. 5D).
Fig. 5
Knockdown of NF-κB inhibits LPS-induced podocyte apoptosis. After transfection of si-Con or si-NF-κB into podocytes for 24 h, NF-κB/p65 (A) and miR-21 (B) were analyzed using western blot and RT-qPCR, respectively. After transfection with si-NF-κB, miR-21 inhibitors or si-NF-κB+miR-21 mimics into LPS-stimulated podocytes, cell ability (C) and cell apoptosis (D) were measured using CCK-8 and TUNEL staining, respectively
*p < 0.05 compared with control group; &p < 0.05 compared with LPS group; $p < 0.05 compared with si-NF-κB group.
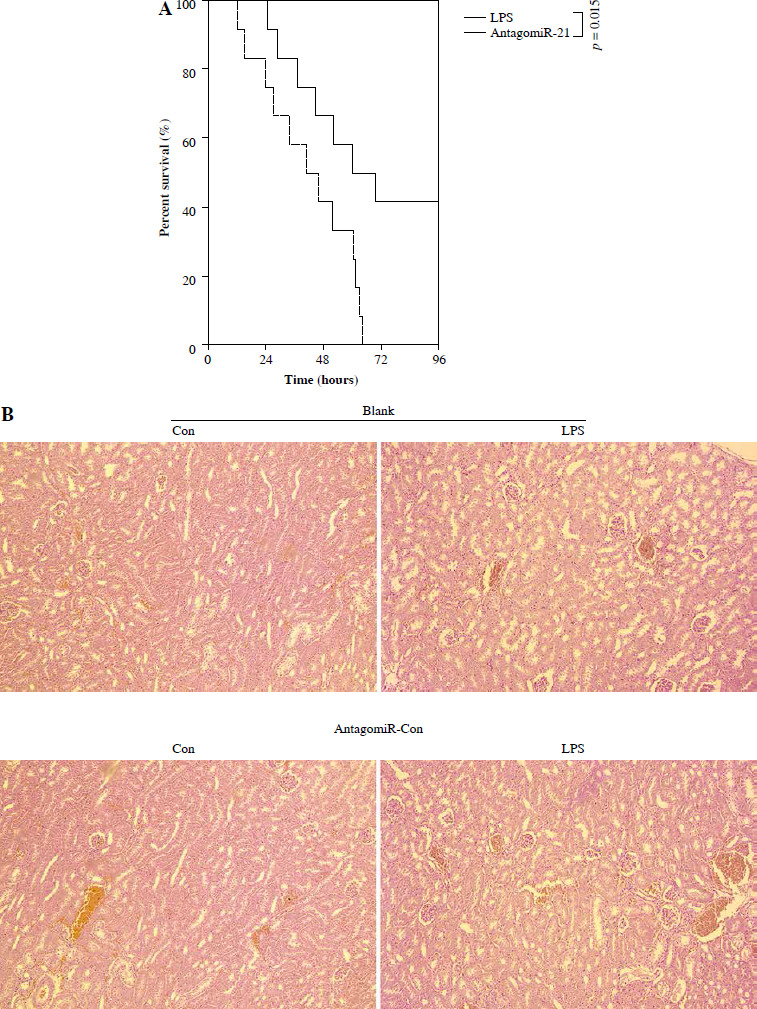
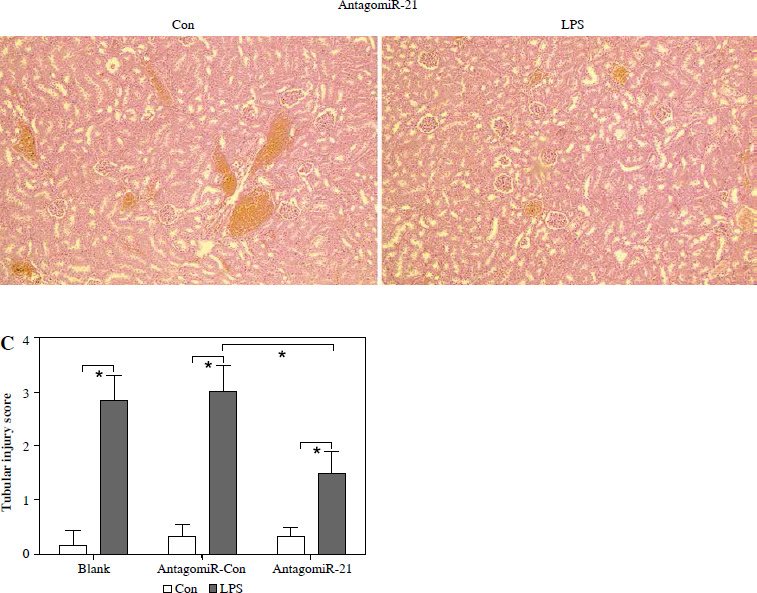
AntagomiR-21 alleviates LPS-induced AKI and the inflammatory response
Based on the aforementioned information, it is evident that the overexpression of miR-21 is associated with LPS-induced AKI. We postulated that the administration of antagomiR-21, an inhibitor of miR-21, could potentially ameliorate LPS-induced AKI. In our experimental study, a dose of 20 nM/0.1 ml of antagomiR-21 was administered via tail-vein injection after a 30-minute LPS treatment. The results depicted in Fig. 6A demonstrate a significant increase in the survival rate (41.7%) of septic mice following antagomiR-21 treatment. Furthermore, the findings from renal histologic examination (Fig. 6B) and tubular injury score assessment (Fig. 6C) provide additional support for the significant improvement in LPS-induced renal injury upon administration of antagomiR-21.
Fig. 6
AntagomiR-21 improves survival rate and alleviates LPS-induced AKI. LPS-treated mice administered antagomiR-21; survival rate was analyzed in the next four days after LPS injection (A). LPS-treated mice administered antagomiR-21; renal histologic examination (B) was performed and tubular injury score (C) was determined in the kidney
*p < 0.05.
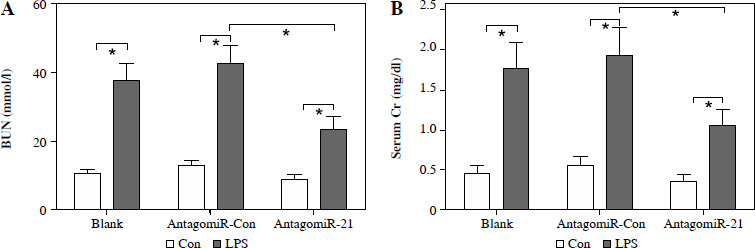
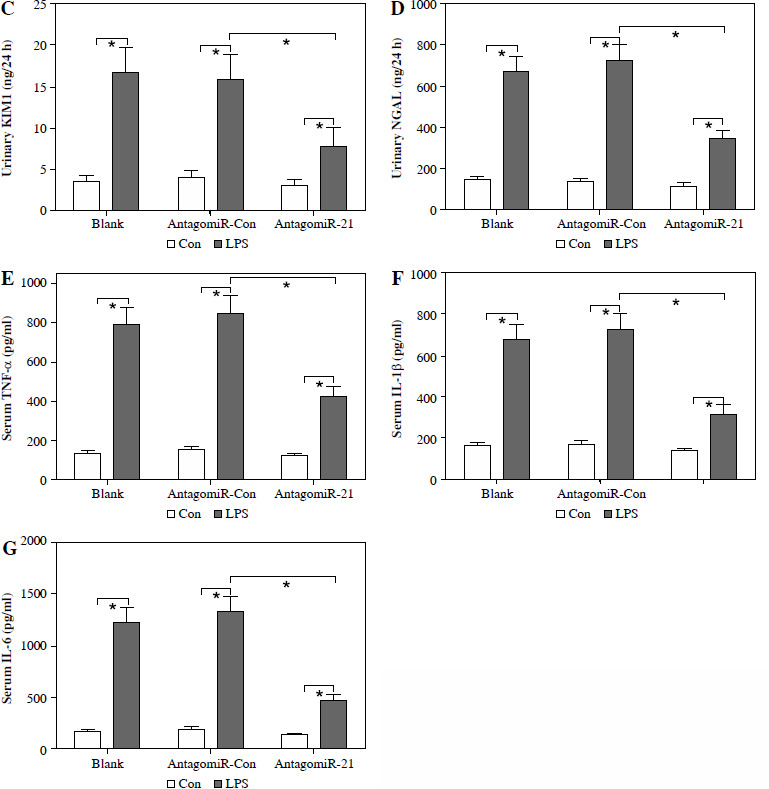
The administration of antagomiR-21 significantly attenuated the up-regulation of BUN (Fig. 7A), serum Cr (Fig. 7B), urinary KIM1 (Fig. 7C), and NGAL (Fig. 7D) induced by LPS. Additionally, antagomiR-21 administra-tion effectively inhibited the LPS-induced increase in serum inflammatory cytokines, namely TNF-α (Fig. 7E), IL-1β (Fig. 7F), and IL-6 (Fig. 7G).
Fig. 7
AntagomiR-21 alleviates LPS-induced inflammatory response. LPS-treated mice administered antagomiR-21; biochemical parameters BUN (A), serum Cr (B) urinary KIM1 (C) and NGAL (D) were detected to evaluate AKI. Inflammatory cytokines TNF-α (E), IL-1β (F) and IL-6 (G) in the serum were evaluated using ELISA assays
*p < 0.05
Discussion
In our study, it was demonstrated that HYP functions as a potential inhibitor of NF-κB, leading to an improvement in the survival rate of septic mice. Additionally, HYP directly blocks LPS-induced podocyte apoptosis and AKI. The mechanism underlying these effects involves the modulation of NF-κB, which in turn affects the transactivation of miR-21. The over-activation of the NF-κB/miR-21 signaling axis has been implicated in LPS-induced podocyte apoptosis and AKI. These findings indicate that HYP acts as a nephroprotective agent in septic mice, and its molecular mechanism involves, at least partially, inhibition of the NF-κB/miR-21 signaling axis.
The study of Novelli et al. showed that HYP has the ability to suppress the expression of NF-κB/p65 in pancreatic β cells when exposed to a cytokine mixture consisting of IFN-γ 400 U/ml, IL-1β 50 U/ml, and TNF-α 150 U/ml [27]. Furthermore, HYP treatment exhibits a protective effect against apoptosis and inflammation by reducing the expression of genes associated with these processes in cytokine mixture-stimulated pancreatic β cells [27]. In pancreatic β cells and in obese or diabetic animal models, HYP mediates diverse signaling pathways, including NF-κB, to prevent diabetes [20, 28]. HYP mitigates β-amyloid protein-induced apoptosis and inflammation in PC12 cells by reducing NF-κB/p65 in a concentration-dependent manner [29]. These findings suggest that NF-κB may be a therapeutic target of HYP in inflammation-related pathological situations. In agreement with previous findings [20, 27-29], the findings of our study indicate that the administration of HYP effectively inhibits the inflammatory response and reduces the expression of NF-κB/p65 in podocytes stimulated with LPS, as well as in mice with sepsis induced by LPS. Furthermore, the knockdown of NF-κB resulted in a decrease in the proportion of apoptotic cells in LPS-stimulated podocytes. In terms of renoprotection, our results demonstrate that HYP administration mitigates renal tubular injury in septic mice.
NF-κB is an inflammation-related mediator, and its over-activation has been associated with the elevation of pro-inflammatory cytokine production [30]. Herein, we investigated an NF-κB-based regulatory network to mediate miR expression. The NF-κB-miRs axis has frequently been reported in inflammation-related diseases, such as arthritis and sepsis [12, 31]. NF-κB/p65 could bind to the promotor of miR-34a to drive its transactivation that contributes to the pathogenesis of arthritis [31]. Zhou et al. highlighted that the NF-κB/p65 subunit bound to the promoter elements of miRNA genes reinforced the transactivation of miR-17-92, miR-125b-1, miR-21, miR-23b-27b-24-1, miR-30b and miR-130a [12]. In addition, NF-κB mediated the lipoteichoic acid-induced inflammatory response in human airway epithelial cells via inhibition of miR-149-5p [32]. In our study, elevated NF-κB/p65 potentiated miR-21 expression in LPS-treated podocytes and septic mice, leading to AKI and an overactive inflammatory response. Lack of miR-21 expression reduced LPS-induced podocyte apoptosis and improved LPS-evoked renal tubular injury. The NF-κB-miR-21 axis creates a positive feedback network controlling the inflammatory response and renal injury in septic models.
As reported previously, overexpression of miR-21-5p accelerates reactive oxygen species production, the inflammatory response and pyroptosis of podocytes [33], and miR-21 antagonism improves renal injury and podocyte dysfunction in in vivo and in vitro diabetic models [34]. In chronic kidney disease patients with glomerular injury, up-regulation of miR-21 is observed in urinary exosomes [35]. These findings suggest that restraint of miR-21 may be a potential therapeutic target to prevent renal injury. A previous study indicated that LPS-induced up-regulation of miR-21 leading to podocyte injury [36]. Consistent with the previous finding [36], miR-21 expression was acutely increased in LPS-treated podocytes and in the kidney of sepsis-treated mice. Importantly, inhibition of miR-21 by HYP or antagomiR-21 alleviated LPS-induced podocyte apoptosis and AKI.