









To the Editors,
We read with great interest the manuscript of Binkowska et al. discussing the recent data on the safer venous thromboembolic (VTE) profile of oestradiol (E2) containing hormone therapies compared to ethinyloestradiol (EE) or conjugated equine oestrogens (CEE) for their use in contraception or in the relief of menopausal symptoms [1]. While we fully agree with the authors about the epidemiological and biological data showing that oestradiol containing pills represent a safer alternative than EE regarding the risk of VTE, the physiopathological mechanism explaining these observations deserves further explanations [2]. The authors pointed out that this is in line with the observations of changes in individual biochemical markers elicited by these treatments [1]. Evaluation of individual markers of coagulation is not predictive of the VTE risk because of the multiple interactions in the coagulation cascade. In contrast, the VTE risk may correlate with the changes in global tests of coagulation which integrate all haemostatic changes [3].
The increased risk of VTE is mainly explained by the impact of oestrogens on the resistance towards activated protein C (APC) and on augmented thrombin generation [4–6]. Our group has recently demonstrated that a higher risk of VTE is observed with pills generating a higher APC resistance, as measured by the endogenous thrombin potential (ETP) based APC resistance test [5, 7, 8]. This has also been observed, although less objectively, by other teams [9]. The correlation between the APC resistance induced by estrogenic therapies and the risk of VTE is not surprising and has been demonstrated for many years [6].
Currently, there are 3 different oestrogens on the market used in combined oral contraceptives: EE, E2 (and its valerate) and estetrol (E4). In their manuscript, Binkowska et al. aimed at discussing the most recent research findings in this area but failed to report E4, this new oestrogenic molecule which has a particular pharmacodynamic and pharmacokinetic (PK) profile [1]. Estetrol is the first natural fetal oestrogen with a selective action in tissues (NEST) [10]. It acts as an agonist for the ER α nuclear receptor but is an antagonist of the membrane initiated steroid signalling (MISS) initiated by E2 [11]. In contrast to the other oestrogens, it is not metabolized by CYP enzymes and does not give rise to carcinogenic nor to biologically active metabolites. These properties translate into clinical benefits such as absence of drug-drug interaction, lower risk of breast cancer and a small impact on the liver (including on lipid, glucose metabolism and haemostasis proteins) [11]. In contraception, E4 15 mg is associated with drospirenone (DRSP) 3 mg [12]. Its haemostatic impact, extensively studied during the clinical development programme [13–15], shows that E4/DRSP has a lower impact on haemostasis than EE in association with either levonorgestrel (LNG) or DRSP [14, 15]. In silico modelling supports the hypothesis that E4/DRSP has a relative risk (RR) of VTE compared to a non-user of around 1.6, which is lower than the RR of 2.2 to 2.4 observed with EE/LNG. This model also highlights the lower risk of VTE with E2/nomegestrol acetate, confirming its robustness to predict the risk of VTE (Fig. 1 A) [7].
Fig. 1
A) Association between normalized activated protein C sensitivity ratio (nAPCsr) data and relative risk of venous thromboembolism in women using combined oral contraceptives; B) mean thrombogram [2.5th–97.5th percentile] of childbearing age women not using any hormonal contraception (baseline population) (n = 86; yellow) and mean thrombogram [95% CI of the mean] after 6 cycles of estetrol 15 mg associated with drospirenone 3 mg (n = 34; pink); C) mean thrombogram [2.5th–97.5th percentile] of untreated postmenopausal women (n = 168; orange) and mean thrombogram [95% CI of the mean] after 12 weeks of estetrol 15 mg (n = 32, red)
CI – confidence interval, COC – combined oral contraceptive, CPA – cyproterone acetate, DNG –dienogest, DRSP – drospirenone, DSG – desogestrel, EE – ethinyloestradiol, E2 – oestradiol, E4 – estetrol, GSD – gestodene, LNG – levonorgestrel, nAPCsr – normalized activated protein C sensitivity ratio, NOMAC – nomegestrol acetate, RR – relative risk, SD – standard deviation, VTE – venous thromboembolism
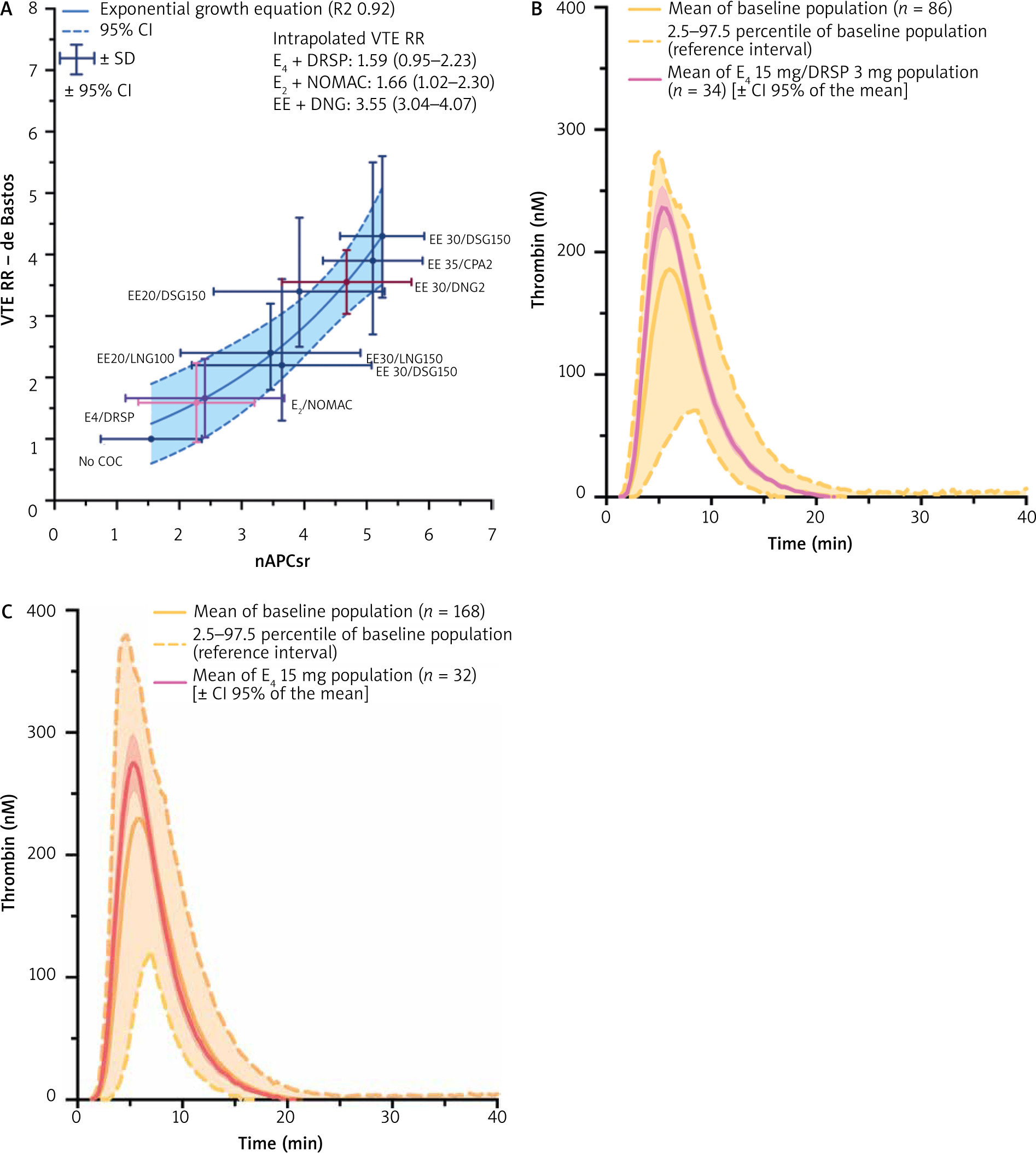
Estetrol is also currently studied for its effective relief of menopausal symptoms [11]. Indeed, for menopausal therapy E4 at the dose of 15 mg daily shows a limited impact on haemostasis parameters [16]. This is very interesting since again in this population, the risk of VTE associated with the use of oestrogen is related to APC resistance induced by hormonal therapies (HT) [17]. According to the data presented by Panay et al. there is a reduced risk of VTE with E2 1 mg/P4 100 mg compared to CEE/medroxyprogesterone acetate [18]. Similar observations were made some years ago by Smith et al., who reported an RR of VTE of 2.08 for CEE compared to E2 [19]. This group also reported that the APC resistance was lower in E2 users compared to CEE users. Thus, in terms of risk of VTE with HT, oral CEE can be categorized as the compound with the highest risk followed by oral E2 and then transdermal E2 [6, 19]. Similar to the observations in the field of contraception, the risk of VTE is also mainly explained by resistance towards APC. The fact that E4 has a similar impact on ETP-based APC resistance as transdermal E2 is reassuring, since this new oestrogen may be classified as the first orally available HT with a low impact on haemostasis [16]. The recent data presented at the 20th World Congress of the International Society of Gynecological Endocrinology and at the 18th World Congress on Menopause of the International Menopause Society confirm this statement. These data show that E4, either associated with DRSP or alone, has a negligible and not clinically relevant impact on thrombin generation, a global coagulation test sensitive to the changes induced by oestrogenic compounds (Fig. 1 B, C) [15, 20]. Added to its low impact on APC resistance and the accumulating evidence that these biological changes are associated with the increased risk of VTE observed in women on HT or combined hormonal contraceptives, E4 represents an oestrogen with an expected low risk of VTE.
The oral intake of E2 generates an unbalanced non-physiological oestrone/oestradiol ratio (E1/E2 = 5) in contrast to that observed in women with endogenous ovarian activity (E1/E2 = 1) [21, 22]. Importantly, during oral treatment, the first liver passage of oestrogens after rapid absorption in the gut is characterized by high local exogenous hepatic steroid levels, which are about 4-fold higher than the peripheral serum concentrations [23]. Moreover, owing to the high permeability of the hepatic microvasculature and the higher availability of protein-bound oestrogen for influx into the liver as compared to other organs [24, 25], the impact on hepatic metabolism of orally applied oestradiol is much higher than that of the transdermal route causing the well-known pro-thrombotic effect [17, 21]. This rapid conversion of E2 into E1 results in a high E1/E2 ratio and contributes to the prothrombotic impact of oral E2. Different groups indeed have shown a correlation between VTE risk and E1 plasma levels [21, 26]. In addition, the carriers of the CYP3A5*1 allele which accelerates the E2 into E1 conversion have a 30-fold higher risk of VTE [26].
In summary, although E2 use appears to be safer than EE or CEE, its oral administration leads to non-physiological, unbalanced metabolite formation, which contributes to the well-known adverse effects of E2 on coagulation. The pharmacodynamic and PK characteristics of E4 (absence of active metabolites such as E3, E2 and E1, absence of hydroxy-metabolites and cancerogenic metabolites) indicate that E4 may represent a safer oestrogen.