






Introduction
Destruction of joints is the main symptom of rheumatoid arthritis (RA). A possible explanation of this process lies in aggressive attachment of synovial fibroblasts to cartilage [1], followed by a release of proteolytic enzymes such as matrix metalloproteinases and serine proteases [2–4].
Psoriatic arthritis (PsA) is a seronegative chronic inflammatory skin and joint disease. It affects metacarpophalangeal and interphalangeal joints of the hands and feet, as well as knees and ankles [5]. Scarce data exist relating the role of hypoxia to the development of PsA [6]. Hypoxia leads to the formation of reactive oxygen species which cause damage to DNA, proteins, and lipids involved in angiogenesis, cell differentiation, migration and proliferation [7].
A high expression of NOX-2 (NADPH oxidase-2) has been found in a study with 54 patients suffering from arthritis (33 with RA and 21 with PsA). A high NOX-2 expression was correlated with low synovial PO2 levels, and with a high expression of vascular endothelial growth factor (VEGF), Ang-2, factor VIII, neural cell adhesion molecule, and α-smooth muscle actin. Moreover, patients treated with anti-tissue necrosis factor (TNF)-α agents showed a decrease in the NOX-2 expression level and an increase in synovial PO2 [8]. Low in vivo oxygen levels have been observed in the synovium of PsA patients. Hypoxia causes an increase of urokinase plasminogen activator (uPA) stimulated release of matrix metalloproteinases (MMPs), and hypoxia-inducible factor (HIF), which in turn induces VEGF production [9].
Synovium of patients suffering from rheumatoid arthritis has a significantly higher expression of components of the plasminogen activation system (PAS) compared with non-arthritic synovium, or from patients with osteoarthritis [10].
The PAS is a proteolytic array of free and membrane bound proteins [11], which modulate the cellular response in terms of mobility, proliferation, viability, and phenotype. Directly, it is responsible for degrading extracellular matrix (ECM) protein [12].
The key protease of PAS is plasmin [13], which is activated through cleavage of plasminogen by uPA in cooperation with the urokinase plasminogen activator receptor (uPAR) [14]. uPA is released from intracellular storage by exposure to cytokines [10, 15] and serum molecules such as interferon γ [16], fibronectin [17], transforming growth factor-β1 [18], and HIF [19] as well as other inflammatory mediators [20, 21]. uPA release and plasminogen conversion activates the proteolytic processes necessary to resolve the source of inflammation [22], trauma [23], or autoimmune fibrosis [24] by stimulating cellular migration [25], proliferation [26], extracellular remodelling [27] and angiogenesis [28]. The effectiveness of uPA is strongly regulated by plasminogen activator inhibitors (PAI-1, PAI-2) and nexin [29]. In brief, uPA and PAI-1 are responsible for modulating the PAS in response to tissue damage under inflammatory, traumatic or autoimmune stimuli [30].
Recently there have been multiple reports that in RA [31] as well as in breast cancer [32] it is the co-expression of PAI-1 with uPA, which has been found to enhance angiogenesis, cellular mobility, invasiveness and excessive ECM turnover. PsA lesions also exhibit increased angiogenesis due to PAI-1 binding to uPA and cell adhesion molecules [33].
Physiological activation of PAS could encourage dedifferentiation under hypoxic [34, 35] and physical stressors [36, 37]. Dedifferentiation is also present with uPA/PAI-1 upregulation in RA [38] and ovarian cancer [39]. PAS functions are also important for growth and development embryologically [40], however are not necessary for survival [41] and come more strongly into play during injury [42]. The adipose tissue is thought to be an adipokine reservoir, which can locally maintain the effects of PsA lesion development [43]. In our previous work the adipocyte response to PAI-1 and uPA co-stimulation has been shown to be pro-angiogenic through a concentration dependent mechanism reacting to hypoxia [44].
Aim
Growth medium starvation and the associated oxygen stress can stimulate human fibroblasts like synoviocytes (HFLS) to switch to heavy catabolic metabolism [45] and low-density lipoprotein (LDL) production [46]. Notably LDL receptor-related protein is crucial for mediating PAI-1 induced cellular migration [47], as it is yet another membrane bound target for PAI-1 colocalization [48].
Reports suggest that a non-catalytic colocalization of uPA to uPAR is sufficient [49–54] for mediation of the uPA associated angiogenesis, proliferation, and mobility seen in arthritic damage. Alternatively uPA needs to colocalise to uPAR and LDR in order to exert these effects [55]. Since colocalization of uPAR to LDR is mediated via PAI-1 [56], we aim to prove that uPA and PAI-1 have an optimum concentration for exerting these effects, and suggest they are each limited by the other when the baseline production is affected by the environmental conditions such as inflammation induced hypoxia.
Material and methods
Cell culture
HFLS were obtained from ScienCell Research Laboratories (Carlsbad, USA). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich, Poland), supplemented with 10% Foetal Bovine Serum (FBS) and an antibiotic-antimycotic mixture (penicillin, streptomycin and amphotericin) to 1% concentration, on 25 cm2 plates at 37°C and 5% CO2. Passaged by washing with Hank’s Balanced Salt solution followed by 0.25% trypsin in 0.53 mM EDTA. After 10 min incubation cells were mechanically detached by striking against the workbench, following which 8 ml of culture medium was used to wash the cells and distribute across two new plates. The medium was changed every 2–3 days until 80% confluence. Cells were saved to library in liquid nitrogen by suspending in 10% DMSO.
UPA, PAI-1, rich and hypoxic medium exposure
Test cultures of HFLS were grown to 80% confluence on small culture flasks in 4 ml growth medium. Passaged to 96 well plates and normalised for 24 h in standard cell culture medium, the cells were subsequently exposed to test media with human recombinant uPA (5, 20, 40 and 80 μg/ml) and PAI-1 (0.1, 1 and 5 μg/ml) (ProSpec-Tany TechnoGene Ltd., Israel) at both rich (10% FBS) and the hypoxic model (0.1% FBS) culture medium for 24, 48 and 72 h. UPA and PAI-1 concentrations were chosen for bioactive levels both under physiological and pathological processes (74–79), as no data for bioactive concentrations in vivo vs. in vitro were available.
Cell viability assay
Cell Counting Kit-8 (CCK8; Sigma-Aldrich) was used to assess the effect of test media on HFLS survival. The protocol calls for 10 μl CCK-8 reagent in each tested well of 96 plates, followed by 1 h incubation at 37°C. A series of blank calibration wells with no growth medium or cells was used for measurement control. Viability was measured by absorption of 450 nm wavelength light by formazan reduced WST-8. Wavelength generation and absorption measurement was carried out with Infinite M200 PRO (TECAN, Switzerland). Formazan is sensitive to necrosis induced leakage of lactate dehydrogenase [57].
Statistical analysis
Data across runs were pooled. 24, 48 and 72 h of incubation were labelled 1, 2 and 3 days of incubation, respectively, for clarity. Means, standard deviations (SD), as well as standard error of means (SEM) were calculated for each test condition. Means were plotted on bar graphs, and SEMs were used for positive and negative errors. Regression models were made for each tested growth medium mixture and analysed using two factor ANOVA for all sample groups to assess variance between rates of change in viability, as well as one way ANOVA for effects within each sample group across all concentrations of PAS components and subsequently all durations of incubation. Post-hoc significance was assessed using two tailed equal variance t-tests for all significant ANOVA sample groups. α = 0.05 - LibreOffice Calc statistical package was used.
Results
Control conditions
Significant variation was found for cell viability between the control media across all days of incubation F(2, 4) = 88.02, p = 0.0005. Incubation in rich control medium (M = 0.55, SD = 0.06) resulted in significantly higher viability than in hypoxic medium (M = 0.17, SD = 0.03) t(2) = 7.6, p = 0.018, or blank (M = 0.12, SD = 0.02) t(2) = 18.3, p = 0.003 (Figure 1). Time of incubation had a significant effect on cell viability for the hypoxic medium only. Incubation in hypoxic medium at 24 h (M = 0.19, SD = 0.04) resulted in significantly higher viability than at 48 h (M = 0.14, SD = 0.01) t(3) = 9.7, p = 0.002, or 72 h (M = 0.15, SD = 0.05) t(3) = 4.5, p = 0.02.

PAI-1 in rich medium
Figure 2 – No significant variance, similar results across all tested concentrations and the control medium.

PAI-1 in poor medium
Incubation on day 2 in hypoxic medium (M = 0.14, SD = 0.01) resulted in significantly lower cell viability than in PAI-1 at 0.1 µg/ml (M = 0.32, SD = 0.02) t(2) = 31.1, p = 0.001, 1 µg/ml (M = 0.31, SD = 0.02) t(3) = 25.8, p = 0.0001 or 5 µg/ml t(3) = 18.80, p = 0.0003 (Figure 3). Incubation on day 2 in PAI-1 0.1 µg/ml (M = 0.32, SD = 0.02) also resulted in significantly higher cell viability than at 5 µg/ml t(2) = 7.39, p = 0.019.

PAI-1 in hypoxic medium
Incubation on day 1 in uPA at 20 ng/ml (M = 0.79, SD = 0.12) resulted in significantly higher cell viability than in rich control medium (M = 0.32, SD = 0.02) t(3) = 15.5, p = 0.0006, or in uPA at 5 ng/ml (M = 0.52, SD = 0.07) t(3) = 4.49, p = 0.02 and 80 ng/ml (M = 0.37, SD = 0.08) t(3) = 4.46, p = 0.021 (Figure 4).
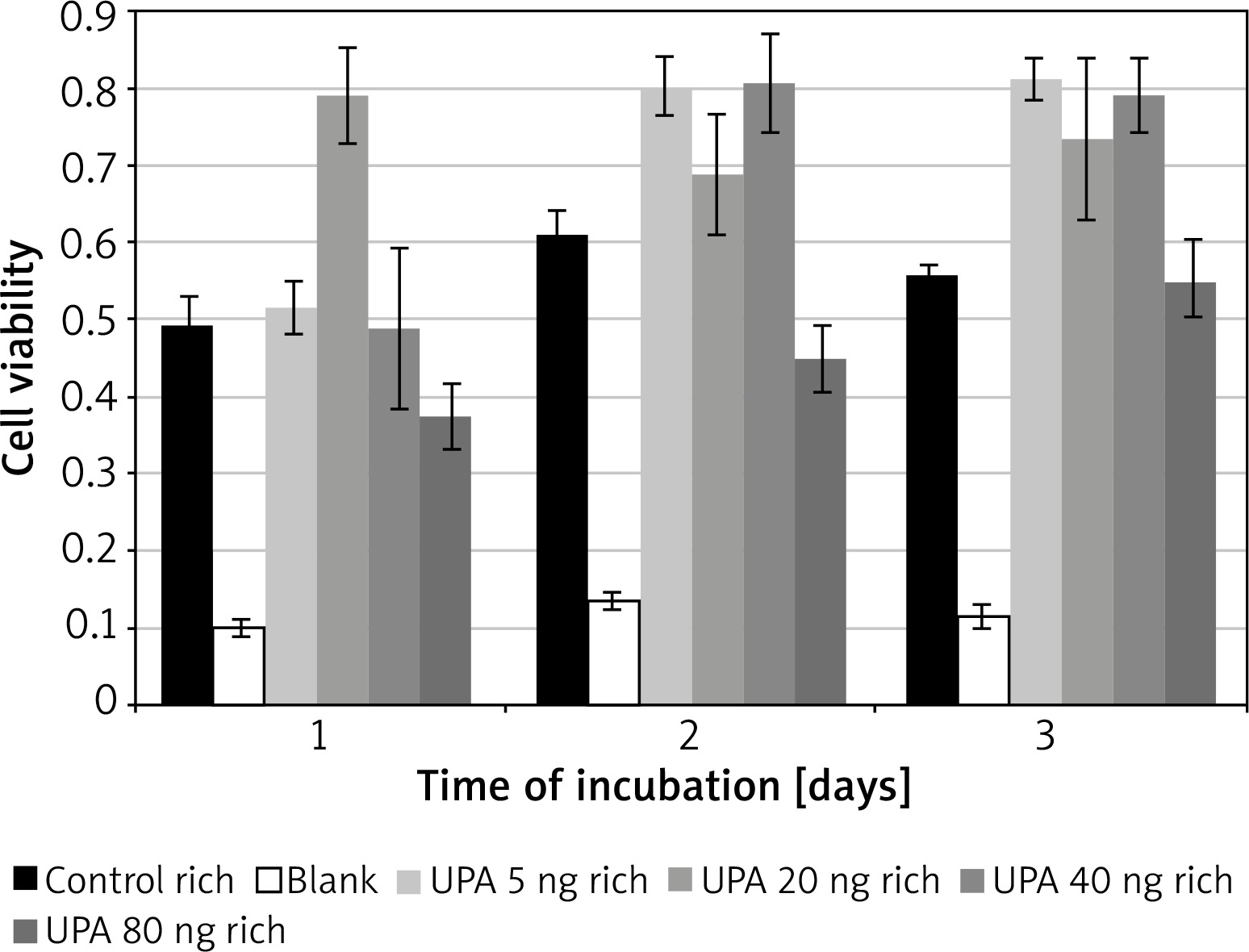
Incubation on day 2 in uPA at 80 ng/ml (M = 0.45, SD = 0.09) resulted in significantly lower cell viability than in rich control medium (M = 0.61, SD = 0.06) t(3) = 3.21, p = 0.049, or in uPA at 5 ng/ml (M = 0.80, SD = 0.08) t(3) = 5.27, p = 0.013 and 40 ng/ml (M = 0.81, SD = 0.13) t(3) = 6.83, p = 0.006.
Incubation on day 3 in rich control medium (M = 0.56, SD = 0.03) resulted in significantly lower cell viability than in uPA at 5 ng/ml (M = 0.81, SD = 0.05) t(3) = 20.64, p = 0.0002, and 40 ng/ml (M = 0.79, SD = 0.09) t(3) = 3.79, p = 0.032.
Incubation in uPA at 5 ng/ml for 1 day (M = 0.52, SD = 0.07) resulted in significantly lower cell viability than for 2 days (M = 0.80, SD = 0.077) t(3) = 6.05, p = 0.009 or 3 days (M = 0.81, SD = 0.052) t(3) = 7.05, p = 0.006.
uPA in hypoxic medium
Incubation on day 2 in hypoxic medium (M = 0.14, SD = 0.01) resulted in significantly lower cell viability than in uPA at 5 ng/ml (M = 0.21, SD = 0.0086) t(3) = 12.08, p = 0.001, 20 ng/ml (M = 0.21, SD = 0.026) t(3) = 5.97, p = 0.009, 40 ng/ml (M = 0.21, SD = 0.015) t(3) = 20.22, p = 0.0003, and 80 ng/ml (M = 0.18, SD = 0.024) t(3) = 6.08, p = 0.009.
Incubation on day 2 in uPA at 40 ng/ml resulted in significantly higher cell viability than in uPA at 80 ng/ml t(3) = 4.86, p = 0.017 (Figure 5).

Discussion
Many pathological processes involve a dysregulation of PAS, such as the upregulation of plasmin activity in arthritis [58, 59] and cancer [60], or its downregulation in fibrosis [61–63]. Cancer therapies utilising PAS suggest stimulating the secretion or delivery of PAI-1, which would stop uPA mediated angiogenesis, however this approach has not brought promising results yet [64]. Connective tissue and autoimmune disorder studies suggest, however, that uPA over activation in RA can be inhibited by PAI-1 suppression [65, 66]. In terms of pathologies which suffer from uPA downregulation, treatment by supplementation of uPA has yielded some positive results in pulmonary [67] and liver [68] fibrosis models, but not in renal fibrosis [69].
Prevalence of either molecule in pathological conditions could stem from testing uPA and PAI-1 in isolation. Their common transcription pathway – Wnt3a/β-catenin induced release of nuclear factor k-light-chain-enhancer of activated B cells (NF-κB) [70–72] – provides further insight. Expression of the urokinase gene can be activated by NF-κB [73]. Inhibition of the Wnt3a/β-catenin pathway has been shown to improve RA outcomes by stimulating synoviocyte apoptosis [74], while PsA associated subchondral degradation is thought to be activated by Wnt5a mediated NF-κB release [75]. This is thought to be due to HFLS overexpression of integrins resulting in overactivation of integrin mediated ECM binding [76], and a higher uPA mediated integrin associated proteolysis and subsequent destruction of those attachments [77–79], resulting in synovial degradation.
Since integrin associated uPA targeting of ECM proteins and subsequent detachment can be mediated by colocalization of PAI-1/integrin/uPAR/uPA [80], a reduced production of PAI-1 by the inhibited Wnt3a/β-catenin pathway could reduce the integrin mediated endocytic recycling [81], resulting in lower reformation of surface integrin attachment and therefore a detachment of the overactive synoviocytes, and their subsequent apoptosis [82, 83]. This phenomenon is further supported by the anti-apoptotic effect PAI-1 has in other cell lines [84–86]. Even in PsA, the effect of anti-rheumatic disease modifying medications as well as more targeted TNF-α inhibitors seems to be heavily dependent on reducing both uPA and PAI-1 together, and not in isolation [87].
In our study we observed the influence of PAI-1 and uPA on HFLS in hypoxia induced by low concentration of nutrients in the culture medium. Incubation in medium containing high serum concentration boosts cell viability, and higher concentrations allow for better cellular adaptation [88]. Presence of nutrients and growth stimulating cytokines is key for cellular survival [89], growth, as well as attachment [76, 90]. In order to mitigate low nutrient stress, human cells cope by producing lipid droplets which initiate fatty acid catabolism [91]. Nutrient stress induced by serum starvation, such as 0.1% FBS, can prime and sensitise cells to apoptotic stimuli [92]. Hypoxia induced by the energy intensive catabolic fat metabolism is such an apoptotic stimulus [93].
In the absence of a phagocytic clearance mechanism, apoptotic cells undergo secondary necrosis releasing internal cell components [94]. These components are immunostimulatory, such as the inflammatory danger associated molecular patterns [95]. The most common systemic phagocytic clearance mechanism is scavenger cells which originate from the myeloid phagocyte system, also known as the professional phagocytes – macrophages [96], which would normally be absent in a HFLS monoculture, as it originates from the mesenchyme [97].
Secondary necrosis in vivo results in an increased inflammatory response and can lead to severely destructive pathology [98]. In vitro, activation of synoviocytes by noxious stimuli in inflammatory conditions can induce a release of latent membrane bound matrix metalloproteinases as well as serine proteases [99]. These proteases, collagenases, and ECM degrading molecules are also released from the cell membrane as a late-stage effect of secondary necrosis, further reducing the attachment of surviving local cultures which lack phagocytic clearance mechanisms. Growth arrest, nutrient depletion, apoptosis and secondary necrosis are all timed events dependent on local nutrient and oxygen availability, as well as clearance of toxic products. We have shown that for HFLS under serum starvation, the completion of these processes could take more than 24 h.
PAI-1 is naturally secreted by synoviocytes during pathological processes such as RA [100], but also serves a physiological role in protecting the synoviocytes from inflammatory processes and modulating the uPA activity [73]. It is elevated during inflammatory processes such as diabetes [101], and reports show that it has an inhibitory function during wound healing [102]. Nonetheless, PAI-1 expression was found to be elevated in invasive breast cancer [103]. Classically PAI-1 inhibits cellular mobility and remodelling which would otherwise be caused by uPA, therefore causing fibrosis and morbidity in obesity and wound healing.
PAI-1 had a significant effect on improving cell viability in hypoxia, especially at low concentrations, with the effect increasing over time. Hypoxia is stress inducing, and the cellular response is catabolic, inflammatory followed by apoptotic and necrotic. Apoptosis is concomitant with formation of blebbing, resulting in detachment internally and externally through fibre contractions [104]. Extracellular PAI-1 could slow down this detachment by inhibiting the uPA, which is secreted in response to apoptosis and necrosis. This observation is in line with other groups finding that PAI-1 can mediate protection against efferocytosis of neutrophils [85], FasL-mediated endothelial cell apoptosis [105], and fibrosis-induced apoptosis of fibroblasts [84]. Since the protective effect is inversely proportional to concentration, suggesting an external limiting factor.
Classically PAI-1 is inhibitory to cellular mobility, survival, and extracellular remodelling. We have seen that in hypoxic conditions, where cellular processes are in a catabolic halt and the apoptotic release of uPA could stimulate ECM detachment, the role of PAI-1 can be different. At low concentrations we suggest it plays a role in disarming the surplus secreted uPA, and inhibiting the apoptotic detachment, increasing proliferation and cell viability. However, at concentrations exceeding the buffering capacity of uPA, PAI-1 would go on to act classically to decrease cell viability. The effect of PAI-1 mediated protection becomes more pronounced over the 3 days, suggesting a level of adaptation of the culture to the hypoxia inducing medium, which could be due to better attachment or, as other groups have found, due to dedifferentiation [106].
uPA is usually defined as a plasminogen activator playing a key role in cell migration, proliferation and ECM remodelling - especially during wound healing [107]. There are many reports of its involvement in pathological processes, most notably cancer, where it has been conducive [108, 109] to invasive phenotypes. Our data also suggest a boost in cell viability at low uPA concentrations.
However, uPA is also involved in stimulating PAI-1 mediated endocytosis [110]. Overactivation of plasmin, and MMP mediated ECM degradation, can cause cellular detachment and stress [111]. If endocytosis of the uPA/PAI-1/uPAR complex exceeds the pace of cellular ability to replenish membrane stocks, cells undergo non-apoptotic death [112]. These alternative modes of interaction with the ECM can serve as an explanation for our observed drop in cell viability at high concentrations of uPA. The resolution of this decrease in viability over the 3 days of incubation could be due to the cellular recycling of excess uPA.
At high uPA concentrations, ECM degradation, proteolysis, and detachment occur too quickly, resulting in cellular stress which maintains the apoptotic state. The hypoxia induced release of uPA could explain why there was no significant difference between the control conditions and the uPA concentrations in hypoxic medium. Increased cell viability over the hypoxic medium on the second day of incubation could be explained by the catabolic hypoxic stress activating the Wnt3a/β-catenin signalling pathway [113], which is found to be involved in apoptotic protection [114] by stimulating both uPA and PAI-1 [70] secretion in tandem, allowing both molecules to follow the classical pathway of increased mobility, proliferation and survival.
Severe and chronic diseases can exhibit overexpression of both uPA and PAI-1, such as in RA or PsA, leading to excessive tissue damage, as well as cases of high PAI-1 cancers metastasizing due to more stable angiogenesis. The localisation of PAI-1 to ECM attachment molecules could prove to be the next step in understanding these interactions. PAI-1 binds to ECM attachment molecules together with uPA/uPAR forming a pentameric complex of uPAR/uPA/PAI-1/vitronectin, which is able to combine further with LDR. The local activation of uPA near PAI-1/vitronectin triggers ECM detachment and uPAR/uPA/PAI-1/vitronectin/LDR clathrin mediated internalization. Homeostatic cellular systems can then designate the clathrin contents to degradation or cell membrane recycling. Elements of this mechanism can be observed during carcinogenesis [115].
Our data suggest that uPA can induce a decrease in cell viability, which in turn could occur by stimulation of detachment and plasmin/MMP activation leading to apoptosis. We have shown that PAI-1 can stimulate cell viability in hypoxic stress conditions. The possibility of stimulating attachment and stabilisation as well as endocytosis via the uPAR/PAI-1/integrin complex, even at low extracellular PAI-1 concentrations, is a known mechanism. We further show that PAI-1 is not inhibitory to cellular proliferation in low stress conditions, while uPA does not stimulate proliferation or protect from apoptosis in hypoxic stress conditions. The inhibitory effect of uPA on cellular viability at high concentrations in rich medium can cause extracellular depletion of PAI-1 which could in turn result in uPA activated detachment and apoptosis instead of PAI-1 mediated endocytosis.
We found that in rich medium both uPA and PAI-1 have a decreasing stimulatory effect on survival with increased dosage, suggesting that outside of hypoxic stress, both of these molecules are homeostatically well regulated by the cell. The introduction of excessive amounts of either molecule overpowers the homeostatic mechanisms resulting in an inhibition of viability. We therefore conclude that the most likely mechanism of excessive, rapid, and destructive tissue turnover as present in PsA is due to the combined activity of both uPA and PAI-1, released by the cell due to the hypoxic stimuli through the activation of NF-κB transcription, depletion of cellular reservoirs, heightened activity of local adipose buffers due to systemic comorbidities and immune responses and homeostatically to buffer the other elements of PAS. Further research will need to focus on optimising these destructive stimuli.