






Introduction
Head and neck squamous cell carcinoma (HNSCC) accounts for the sixth most prevalent cancer worldwide. Oral squamous cell carcinoma (OSCC) is one of the most common malignancies of the head and neck, which arises from the mucosal epithelium of the oral cavity and oropharynx [1, 2]. Although several diagnosis and treatment approaches have been innovated, the incidence of OSCC appears to be increasing worldwide. In addition, the 5-year overall survival rate remains at approximately 50–60% [3, 4]. The treatment strategy of OSCCs depends mainly on conventional therapeutic approaches, such as surgery, radiotherapy, and chemotherapy [5]. Notably, those treatment modalities cause harmful side effects because of their cytotoxic effects on normal cells. Consequently, researchers have been developing anticancer therapeutics that have selective cytotoxicity on neoplastic cells [6].
Nowadays, significant attention has been focused on the use of natural compounds and dietary agents for the prevention and treatment of a wide variety of diseases including cancer. Interestingly, more than 60% of approved drugs are based on natural products, and approximately 75% of the novel anticancer drugs are analogues of phytochemicals or originated from plant-based metabolites [7]. Natural products, especially phytochemicals, have also been investigated extensively for their safe and powerful anticancer therapeutic effects [8]. It has been reported that phytochemical compounds can efficiently kill fast proliferating cancer cells, through modulation of the growth signals, suppression of angiogenesis, stimulation of apoptosis, as well as inhibition of metastasis [9, 10]. In addition, many phytochemical compounds have shown chemo-preventive effects on HNSCC, such as green tea, curcumin, and lycopene [6]. The chemo-preventive action might be attributed to several antioxidants, anti-inflammatory, anti-mutagenic, and epigenetic modulators [11].
Coffee is one of the most commonly consumed beverages globally, which has been closely pertinent to human health. It is a complex chemical mixture of more than 1000 diverse phytochemical constituents including polyphenols, caffeine, chlorogenic acid (CGA), and diterpenes [12]. Being rich in natural antioxidants, it has an outstanding potential to be utilised as a nutraceutical agent in various medicinal applications [13]. Research studies have revealed the potential effects of coffee consumption on several diseases, such as diabetes, obesity, cardiovascular, and neurodegenerative diseases, in addition to cancer [14, 15]. It was demonstrated that coffee consumption can mitigate the risk of several types of cancer, such as oral, pharynx cancer, colon cancer, liver cancer, and prostate cancer [16–19]. Studies including meta- analysis and cohort studies have revealed the preventive effect of coffee against OSCC [18, 20, 21].
Coffee is not always consumed in its natural green form, but after different processing procedures such as roasting and decaffeination that alter the concentration of coffee constituents. Caffeine was reported to have potent anti-inflammatory effects through various mechanisms including modifications of cell signalling molecule production, potentiation of the release of anti-inflammatory cytokines, and suppression of the release of pro-inflammatory cytokines, which play a central role in the initiation and progression of autoimmune diseases [22, 23]. The anti-inflammatory effects of caffeine are attributed to its potent antioxidant capability, which results from the high levels of glutathione that scavenge hydroxyl radicals [24]. Furthermore, caffeine was also found to regulate the epithelial-mesenchymal transition and inactivation of the extracellular signal-regulated kinase as well as activation of p38 mitogen-activated protein kinase [25, 26]. It exerts an anti-angiogenic activity via modulation of caspase-3 and B-cell lymphoma 2, in addition to induction of endothelial cell apoptosis [27]. Additionally, it was reported that caffeine inhibits the expression of vascular endothelial growth factor and epidermal growth factor receptor (EGFR) in HeLa-cells and decreases human fibroblast growth factor 19 mRNA levels in colorectal adenocarcinoma cell lines LS174T [26, 28].
Herein, we chemically analysed 3 different types of coffee extracts, unroasted green coffee (GC), medium-roasted coffee (MRC), and decaffeinated coffee (DC). We then measured the concentration of caffeine and other phytochemical constituents, to which the cytotoxic effects were correlated. Roasting is the process of heating GC beans to form volatile organic compounds and develop the complex flavours of coffee [29, 30]. It is believed that roasting coffee generally results in a significant change in the concentration of coffee constituents and consequently the anticancer properties [31, 32]. Nevertheless, it is believed that the decaffeination process, which includes the removal of a substantial amount of caffeine, reduces the anticancer effect of coffee [33, 34]. The cytotoxic effects of the 3 extracts were evaluated against tongue squamous cell carcinoma (OSCC-25) cell lines. The mechanisms of cytotoxicity were also studied by assessing the activity of caspase-3, caspase-9, and EGFR tyrosine kinase (EGFR-TK).
Material and methods
Green coffee beans (750 g) were purchased from local Egyptian markets and divided equally into 3 groups. First group was used in its green form while the rest was further processed to obtain MRC and DC. Tongue squamous cell carcinoma (OSCC-25) cell lines were purchased from the American type culture collection through VACSERA, Egypt. It was stored in liquid nitrogen containers at (–196°C). The reagents for cell culture were purchased from Invitrogen/Life Technologies and Sigma. The entire procedure was carried out in the Tissue Culture Laboratory of the Research and Development sector, VACSERA, Cairo, Egypt, according to the manufacturer’s instructions (Invitrogen/Life Technologies). Two antibodies were used in the present study for the analysis of cell cycle and apoptosis, namely caspase-3 (active) human enzyme-linked immunosorbent assay (ELISA) kit (Catalogue # KHO1091) from Invitrogen Corporation and Human caspase-9 ELISA kit (EIA-4860) from DRG International Inc., USA. EGFR Kinase Assay Kit was purchased from Bioscience (Catalog # 40321).
Roasting process
A total of 250 g of green robusta coffee beans were roasted using an in-house roaster (apparatus brand Primo Roasting Eq.) The initial temperature within roasting drum was 215°C, and then the beans were heated at 228°C for 40 min. The beans were then allowed to cool, vacuum sealed, and stored in a cool dry place until ready for use.
Decaffeination process
The SWISS water decaffeination technique was used to produce DC. First, GC beans were boiled for 3 hours at 80°C to obtain green coffee extract (GCE) solution. The green coffee beans were removed and dried to a moisture content of 25% and the obtained GCE solution was stirred with 10% activated charcoal and left for 1 hour at room temperature. By the end of the previous step, a caffeine-rich-flavour-charged GCE solution was obtained. The dried GC beans were added to the obtained GCE solution and boiled for 3 hours again. This allows caffeine to migrate from GC beans into GCE solution through gradient difference pressure. Green coffee beans were removed from GCE solution and were allowed to dry to a water content of less than 12%. The process was repeated to obtain the desired DC beans. The decaffeinated coffee beans were vacuum sealed and stored in a cool dry place until ready to use.
Preparation of coffee extracts
Green coffee, MRC, and DC powders (50 g) were packed in a Whatmann filter paper and extracted with ethanol using Soxhlet apparatus. The prepared extract was then concentrated under reduced pressure, dried, and stored at 4ºC for further use. A stock solution of each extract (1 mg/ml) was prepared with dimethyl sulfoxide (DMSO) and diluted to the required concentrations with phosphate buffered saline before use.
High-performance liquid chromatography analysis
A serial dilution of caffeine standard was prepared by dissolving standard caffeine in water to yield concentrations of 100–400 µg/ml. Chlorogenic acid standard was also dissolved in water yielding a series of standard CGA solutions with concentrations ranging 100–400 µg/ml. The samples were prepared by dissolving 3 mg of each extract in 1 ml of water. The mixtures were then filtered through a 0.45 µ membrane syringe filter, and 100 µl of each was obtained to be used in high-performance liquid chromatography (HPLC) analysis.
The high-performance liquid chromatography system utilised a Cyano column (4.6 x 250 mm) with a pore size of 5 µm. Caffeine and CGA were analysed using the mobile phase (A: 0.1% trifluoroacetic acid and B: acetonitrile) with a 9-min gradient elution mode (95% A at 0 min, 90% A at 6 min, 95% A at 7 min and 95% A at 9 min) at a flow rate of 1.5 ml/min and ambient temperature (300°C). Caffeine and CGA were detected at wavelengths 270 nm and 324 nm, respectively.
Cell cultures
OSCC-25 cell lines were cultured in Dulbecco’s modified eagle medium supplemented with 10% fetal bovine serum (Hyclone 10 ug/ml of insulin) plus 1% penicillin-streptomycin in T25 cm2 flasks at 37°C in a 5% CO2 incubator. The culture flasks were examined under an inverted phase contrast microscope to assure the viability, sterility, and adequacy of cultured cells. OSCC-25 cell line was sub-cultured to obtain 4 study groups and one negative control (untreated cells), and 3 were treated by the extracts in a dose-dependent manner. The cell viability was determined using the sulforhodamine B (SRB) viability assay.
Sulforhodamine B assay
Cell viability was assessed by SRB assay. Aliquots of 100 µl cell suspension (5 × 103 cells) were placed in 96-well plates and incubated in complete media for 24 hours. Cells were treated with another aliquot of 100 µl of GC, MRC, and DC extracts at various concentrations ranging 10–100 µg/ml. After 72 hours of exposure to coffee extracts, cells were fixed by replacing media with 150 µl of 10% trichloroacetic acid and incubated at 4°C for 1 hour. The trichloroacetic acid solution was removed, and the cells were washed 5 times with distilled water. Aliquots of 70 µl of rhodamine B solution (0.4% w/v) were added and incubated in a dark place at room temperature for 10 min. The plates were washed 3 times with 1% acetic acid and allowed to air-dry overnight. Then, 150 µl of tris(hydroxymethyl)aminomethane (10 mM) was added to dissolve protein-bound SRB stain; the absorbance was measured at 540 nm using a BMG LABTECH®-FLUOSTAR OMEGA microplate reader (ORTENBERG, Germany).
Caspase-9 activity assay
ELISA assay for caspase-3 activity was carried out according to the manufacturer’s instructions. Briefly, 100 µl of the cell lysates was incubated in the microwell plate. The prepared human caspase-9 standard (100 µl) was added to the standard wells (positive control). The sample diluent (50 µl) was added to each sample well, and 100 µl was added to the blank wells (negative control). The coffee extract (50 µl) was pipetted into the sample wells, and the detection antibody (50 µl) was then added to all wells and incubated at room temperature for 2 hours. The antibody solution was then aspirated and the microwell strips were washed 3 times. The diluted anti-rabbit-IgG-HRP (100 µl) was then added to all wells and incubated at room temperature for 1 hour. After removal of the anti-rabbit-IgG-HRP solution, the microwell strips were washed 3 times, and 100 µl tetramethylbenzidine was then pipetted into the wells and the microwell strips were incubated for 10 min. Once the dark blue colour development was confirmed by an ELISA reader at 450 nm, 100 µl of stop solution was pipetted into each well to stop the enzyme reaction. The absorbance was then determined at 450 nm for the sample and the standard wells using a ROBINK P2000 ELISA reader.
Epidermal growth factor receptor tyrosine kinase activity analysis
The proliferative activity in the study groups was detected by measuring EGFR-TK activity using an EGFR-TK activity kit supplied with a detection reagent Kinase-Glo® MAX and measured by BMG LABTECH®-FLUOSTAR OMEGA microplate reader (ORTENBERG, Germany)
In vitro profiling of GC, MRC, and DC extracts was performed to assess their inhibitory activity against EGFR-TK and compared to dasatinib (multi-kinase inhibitor) as a positive control. First, IC50 for EGFR-TK of each extract in addition to dasatinib was applied to each well. The kinase activity data were then calculated by obtaining the percentage of remaining enzyme activity after subtracting the enzyme inhibitory activity of DMSO control reactions as background (Table 1).
Statistical analysis
Data were coded and entered using the statistical package SPSS version 25. Data were summarised using mean and standard deviation for quantitative variables. Significance was determined using analysis of variance for multiple comparisons and post hoc Tukey test for comparison between 2 groups. P-values ≤ 0.05 were considered statistically significant.
Results
Assessment of caffeine content by high-performance liquid chromatography analysis
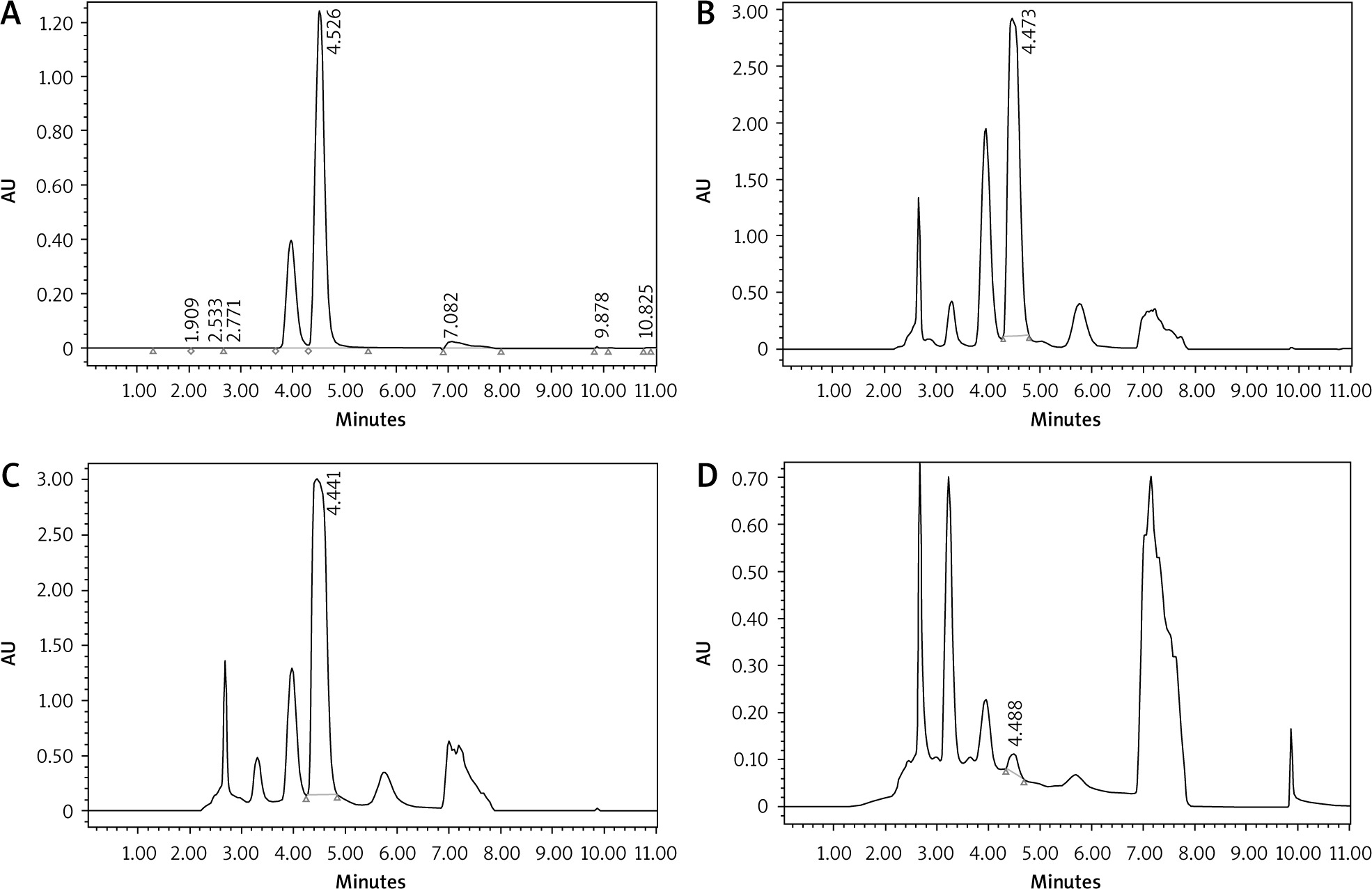
The caffeine content of the 3 extracts (GC, MRC, and DC) was qualitatively analysed with HPLC by comparing the retention time (tR) of the standard caffeine (pure compound) to that of each extract. It is shown in Figure 1 that the average standard caffeine tR was 4.528 min and the mean tR of GC, MRC, and DC extracts were 4.473 min, 4.441 min, and 4.488 min, respectively. Contingent upon the previous results, it is clear that retention times of standard caffeine and the 3 samples are very close, but the caffeine chromatographic peaks have different areas, indicating that the extracts have different caffeine content that should be quantified.
Fig. 1
The chromatograms of standard caffeine (A), green coffee (B), medium-roasted coffee (C), and decaffeinated coffee (D)
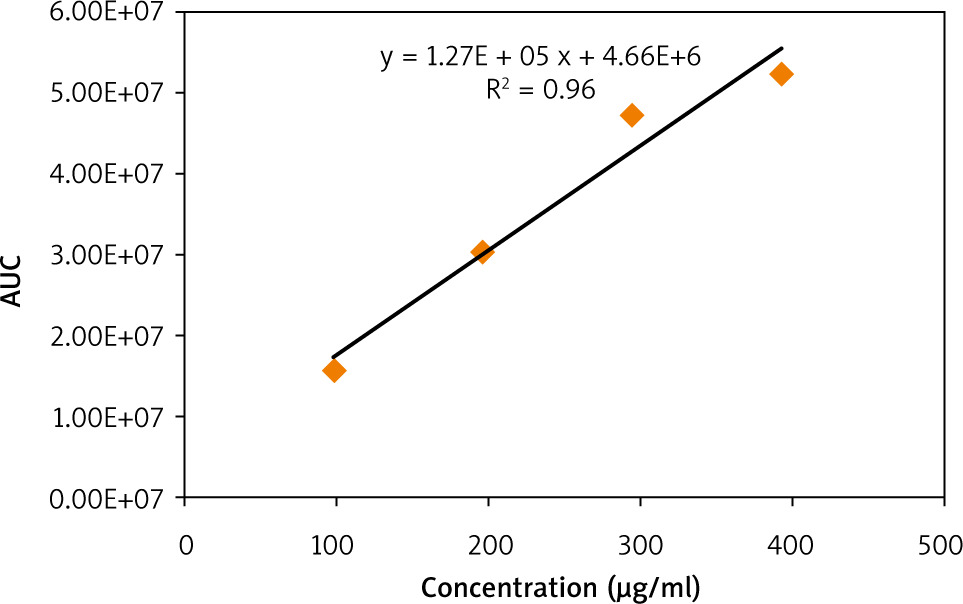
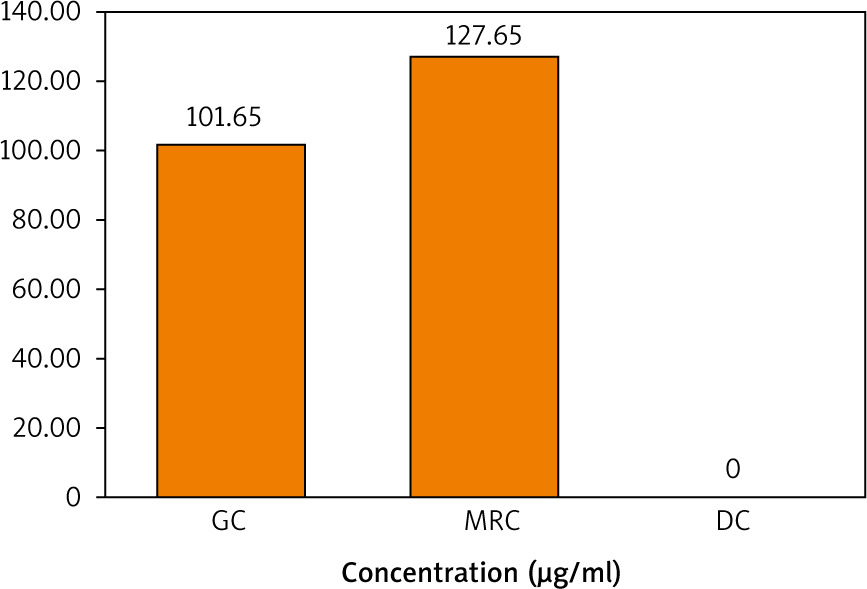
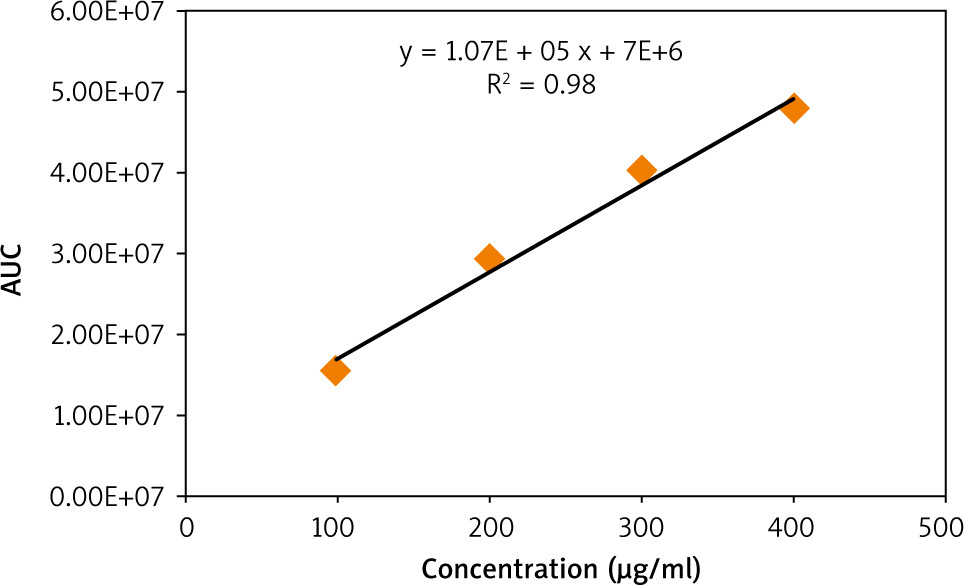
A calibration curve of standard caffeine was then prepared by plotting its concentration (µg/ml) vs. peak areas (Fig. 2). A linear relationship with an R2 value of 0.96 indicates the validity of the calibration curve. The calibration curve was then used to quantify the caffeine content in the 3 extracts. It is demonstrated in Figure 3 that the concentration of caffeine in MRC extract is higher than that in GC, while DC is totally deprived of caffeine.
Assessment of chlorogenic acid content by high-performance liquid chromatography analysis
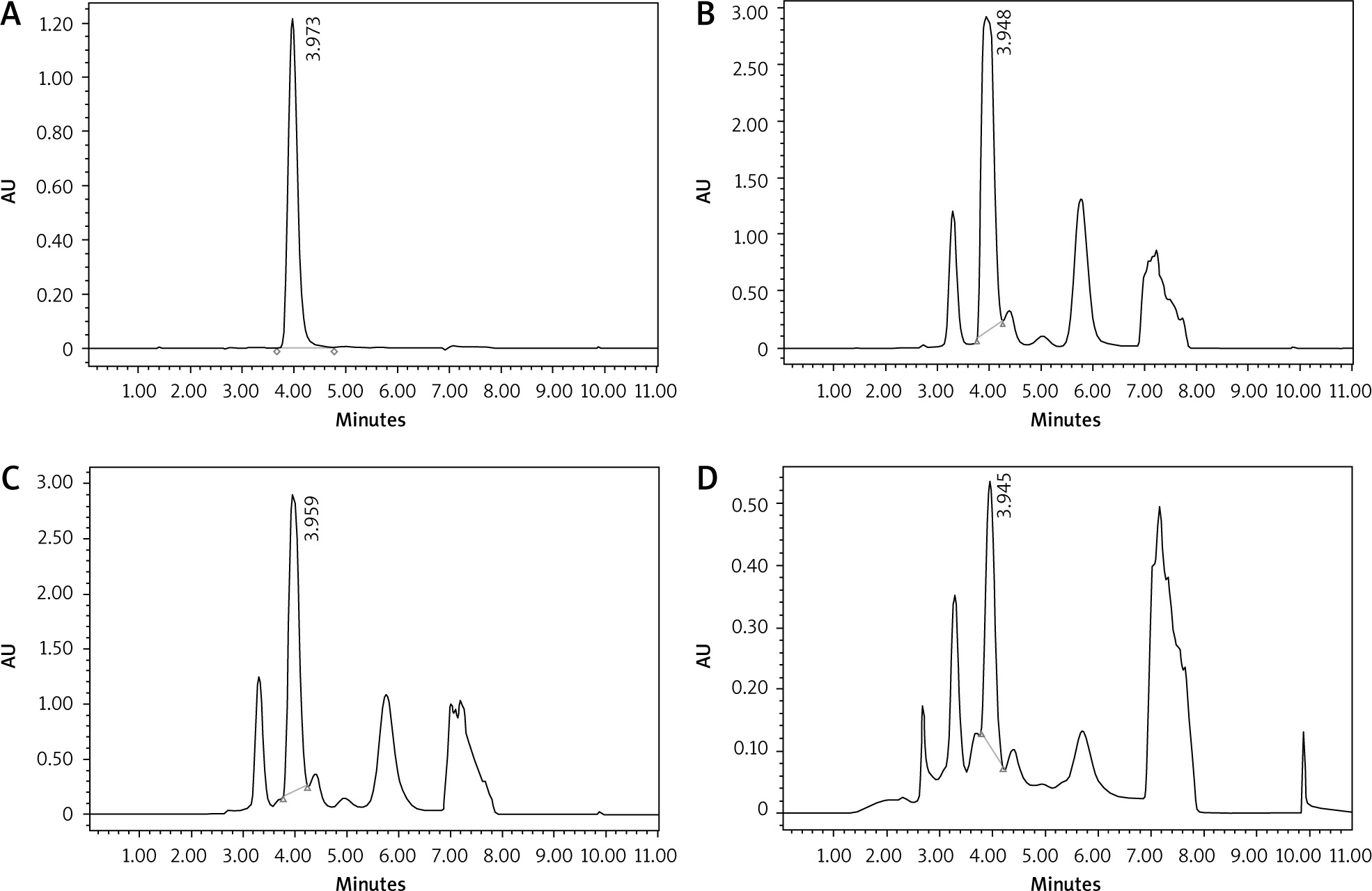
As previously mentioned for caffeine, CGA was qualitatively analysed by comparing the tR of the CGA standard with each extract using the HPLC system. The high-performance liquid chromatography chromatogram of CGA standard, GC, MRC, and DC extracts are shown in Figure 4. It is demonstrated that the average tR for the CGA standard was 3.973 while the other tRs were 3.948, 3.959, and 3.945 for GC, MRC, and DC, respectively. Based on the aforementioned results, it is inferred that the 3 samples have reasonable CGA content. The chromatographic peak areas of CGA in the 3 samples were then calculated for quantification of the CGA content.
Fig. 4
The chromatograms of standard chlorogenic acid (A), green coffee (B), medium-roasted coffee (C), and decaffeinated coffee (D)
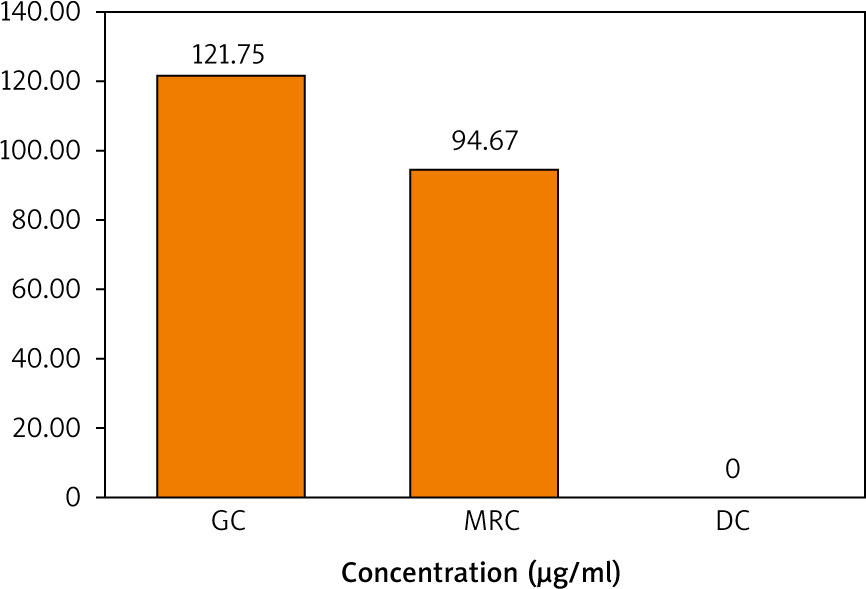
The chlorogenic acid content in the 3 extracts was then quantified using the prepared calibration curve of the standard CGA shown in Figure 5. It is demonstrated in Figure 6 that the concentration of CGA in the GC sample was higher than that in MRC, while DC did not contain any traces of CGA.
Assessment of cell cytotoxicity using sulforhodamine B assay
The cytotoxic effect of GC, MRC, and DC extracts on OSCC-25 cell lines was assessed using SRB assay. The results showed that GC, MRC, and DC extracts induced cytotoxicity upon incubation with the OSCC-25 cancer cell line in a dose-dependent manner. The IC50 values of the extracts were calculated. It was found that GC extract has the lowest IC50 value (16.2 ±0.54) followed by MRC, which has an IC50 value of 56.87 ±2.16, then DC with 104.12 ±4.32, indicating that GC has the highest cytotoxic potential (Table 2).
Assessment of caspase-3 and caspase-9 activity using ELISA
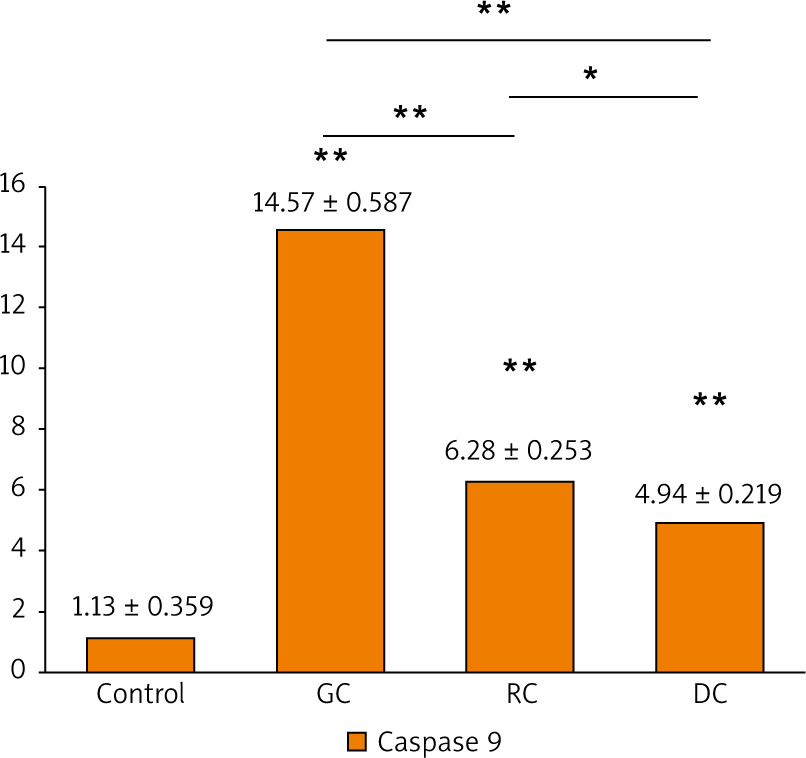
The activation of caspase-3 and caspase-9 proteins was evaluated as an indicator for the apoptosis induced by the extracts. It is shown that there was a statistically significant increase in caspase-3 and caspase-9 concentrations in the GC, MRC, and DC-treated study groups when compared to the control (Figs. 7, 8). It is found that the concentration of caspase-3 was the highest in GC-treated groups (362.4 ±4.67 pg/ml), followed by MRC (233.7 ±3.41 pg/ml), and then DC groups (54.24 ±2.18 pg/ml). Furthermore, GC-treated groups also had the highest concentration of caspase-9 (14.57 ±0.587 ng/ml), followed by MRC and DC, with concentrations of 6.283 ±0.253 ng/ml and 4.942 ±0.219 ng/ml, respectively.
Fig. 7
The bar chart illustrating the concentration of caspase-3 proteins (pg/ml) in OSCC-25 cells in each study group
Data are represented as mean ± standard deviation. The significance was tested using one-way analysis of variance and post hoc analysis. * p < 0.05 (significant) ** p < 0.001 (highly significant)
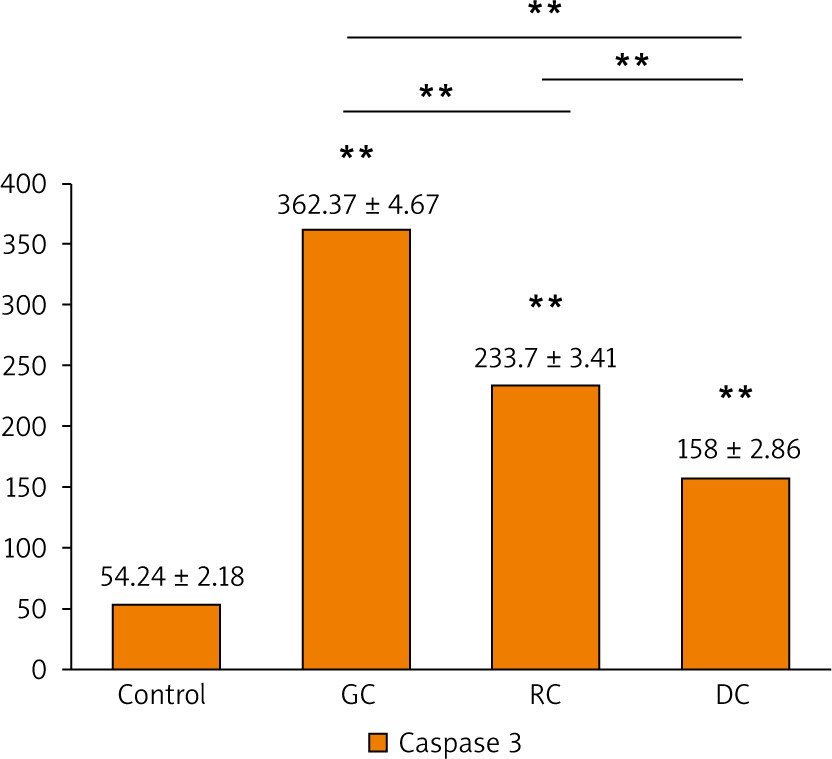
Fig. 8
The bar chart illustrating the concentration of caspase-9 proteins (ng/ml) in OSCC-25 cells in each study group
Data are represented as mean ± standard deviation. The significance was tested using one-way analysis of variance and post hoc analysis. * p < 0.05 (significant) ** p < 0.001 (highly significant)
Assessment of epidermal growth factor receptor tyrosine kinase activity
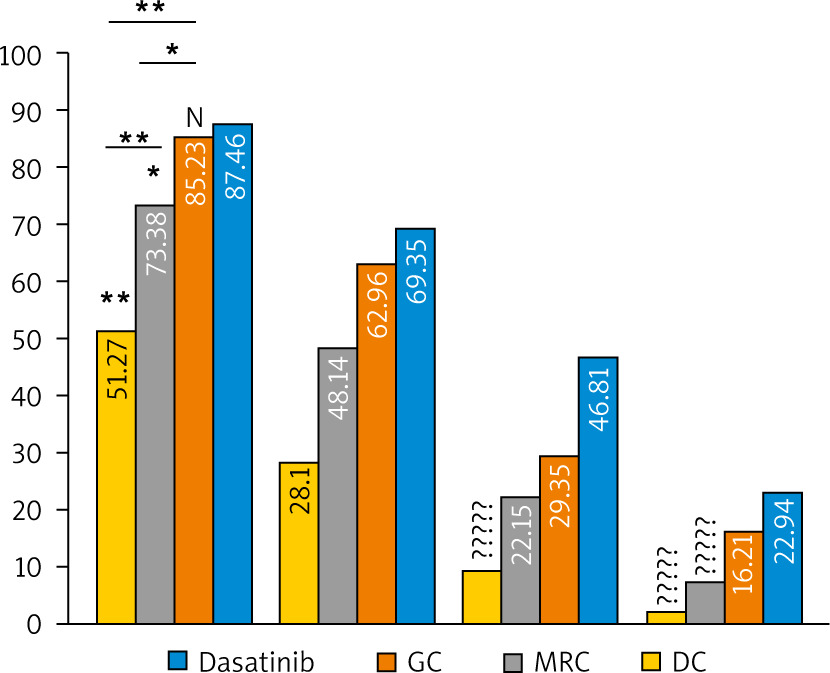
The main EGFR-TK inhibitory effect of GC, MRC, DC, and dasatinib (positive control) on OSCC-25 cell lines was assessed at different concentrations (0.01, 0.1, 1.0, 10 µM). All extracts exert an inhibitory effect on EGFR-TK activity with different IC50 values. The inhibitory effect of the extracts on EGFR-TK activity was directly proportional to the extract concentration. The main percentages of EGFR-TK inhibition by the extracts compared to dasatinib at different concentrations are shown in Figure 9. It is found that GC and dasatinib had no statistically significant difference in EGFR-TK inhibition (87.03 ±4.43 and 85.02 ±5.71, respectively, at 10 µM). Nevertheless, the MRC extract showed a significantly different inhibition from GC and dasatinib (73.47 ±3.99 at 10 µM), while DC had a highly significant difference from all study groups (51.33 ±1.85 at 10 µM).
Fig. 9
The bar chart illustrating the main percentage of epidermal growth factor receptor tyrosine kinase inhibition by green coffee, medium-roasted coffee, and decaffeinated coffee in comparison to dasatinib (control) in OSCC-25 cell lines at different concentrations
The significance was tested using one-way analysis of variance and post hoc analysis. * p < 0.05 (significant) ** p < 0.001 (highly significant), N (non-significant)
Discussion
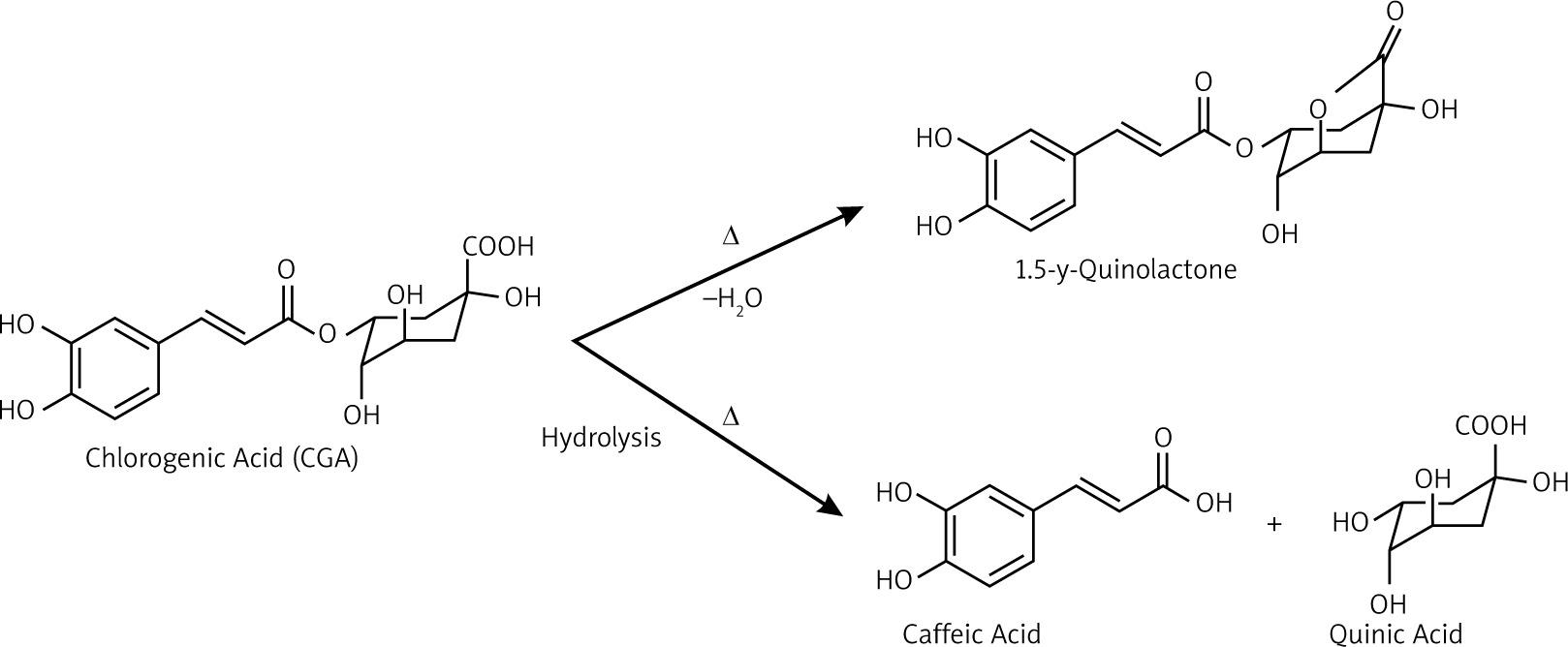
The chemical analysis of GC, MRC, and DC extracts revealed that GC extract has the highest CGA among the samples, followed by MRC, then DC. Nevertheless, the caffeine content was higher in the MRC extract than in GC. Those results can be explained in terms of physical changes and chemical reactions that occur upon roasting. Roasting is usually carried out by heating the coffee beans at 160–260°C, leading to loss of moisture, and thus the concentration of caffeine increases in roasted coffee [35]. However, roasting results in the degradation of CGA into potent antioxidants [36, 37]. Chlorogenic acid undergoes degradation upon roasting through a series of chemical reactions, such as dehydration with intramolecular lactone formation [38], and hydrolysis of the ester bond, forming quinic acid and caffeic acid [39], as shown in Figure 10. Consequently, the CGA content in MRC extract decreases when compared to GC.
The decaffeination process results in the removal of caffeine content from coffee beans. It is usually carried out by several methods, such as water decaffeination and extraction of caffeine using organic solvents, such as methylene chloride and trichloroethylene [40]. Using those solvents during the decaffeination process may result in the extraction of CGA and some phenolic compounds, leading to a lower content of CGA in DC than in GC and MRC.
By investigating the cytotoxicity and apoptotic proteins (caspase-3 and caspase-9) expressions in GC-, MRC-, and DC-treated OSCC-25 cell lines, GC, MRC, as well as DC induced significant cytotoxicity and up-regu-lation of caspase-3 and caspase-9, indicating an apoptosis-induced effect of coffee extracts. The concentration of caspase-3 and -9 was much higher in GC- than in MRC-treated cells because of the higher concentration of CGA. However, the increased caffeine concentration in MRC extract caused by roasting accounts for its substantial cytotoxicity and up-regulation of apoptotic proteins. As expected, DC showed the lowest cytotoxicity and concentration of both caspases when compared to MRC and GC as a result of the depletion of caffeine and CGA. Nonetheless, DC shows considerable cytotoxicity and induction of apoptosis that depend mainly on the total phenolic content that was reported in [34].
One of the main causes behind the uncontrolled growth of tumour cells is an overexpression or self-activation of EGFR [41]. It was found that EGFR is highly expressed in OSCC, and its expression is associated with poor outcomes [42]. Thus, the potentiality of GC, MRC, and DC to inhibit the EGFR-TK activity was measured, and a pronounced growth inhibitory effect by all study groups was reported. In comparison with the OSCC-25 cell group treated with dasatinib, a chemotherapeutic drug targeting the EGFR-TK, the results showed no significant difference between GC and dasatinib, indicating that GC exerts the same inhibitory effect as dasatinib. Moreover, MRC showed a significant potential inhibitory effect on EGFR-TK, which is higher than DC. Those results can be explained in terms of the caffeine and CGA, as previously mentioned.
Conclusions
Coffee is a viable source of natural constituents, such as caffeine and CGA, which are responsible for its cytotoxic effect against OSCC cell lines. It exerts its anti-angiogenic activity through induction of apoptosis by activation of caspase-3 and caspase-9 and inhibition of cell proliferation through inhibition of EGFR. In accordance with our results, it is explicitly clear that GC harnesses its higher concentration of CGA to exert a more potent anticancer effect than MRC. Moreover, MRC loses part of its CGA content, which degrades upon roasting, and therefore, its anticancer effect is mitigated despite its higher caffeine concentration. On the other hand, DC had the least anticancer effect because of decreased concentration of caffeine and CGA upon decaffeination.
Overall, based on our data and the previous research works, our study underlined that it is quite clear that coffee, especially GC, has important anticancer potentials against OSCC, making it a promising chemo-preventive and chemotherapeutic agent for oral cancer treatment; this is likely to be confirmed by further investigations. This study provides supportive data for future investigations that will lead to their use in cancer therapy.